- A+
复合吲哚合成中的冒险:( - ) - Fischerindole I,(+) - Fischerindole G和(+) - Weltwitindolinone A
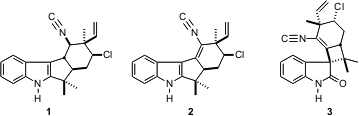
斯克里普斯拉霍亚的菲尔S.巴兰已经描述了(J.化学会志。 2005,127,15394. DOI:10.1021 / ja056171r)优雅的条目添加到复杂的吲哚衍生的天然产物( - ) - fischerindole G(1),(+) - fischerindole I(2)和(+) - weltwitindolinone A(3)。
制备1和2的起点是R-香芹酮的环氧化物4。烯醇化后用乙烯基溴化镁5打开,得到醇6。该反应的产率是适中的,但它直接提供6,所有非吲哚碳骨架的1,适当地被氧化,呈对映体纯的形式。
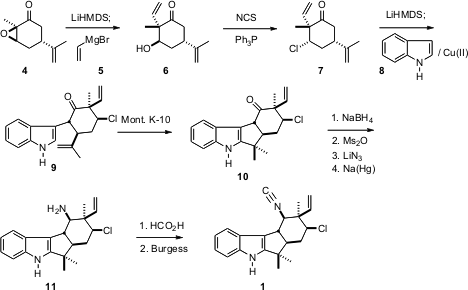
值得注意的是,受阻的仲环己醇的氯化干净地进行,得到7。 7的烯醇化物与吲哚8的氧化缩合得到关键中间体9。经过一些实验,发现粘土蒙脱石K-10会使环化效应达到10。还没有将环己酮转化为赤道胺的一般还原胺化方法,因此将10 还原成轴向醇,将其转化为赤道叠氮化物。然后选择性还原,得到胺11,其甲酰胺脱水得到1。生物碱1 如此制备的是天然产物的对映体。
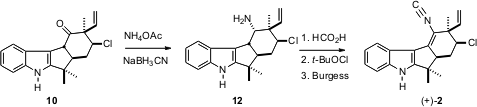
不可能将1或前体甲酰胺脱氢成 2。因此,酮10选择性地还原为轴向胺12。将衍生的甲酰胺暴露于t- BuOCl,然后将硅胶和Et 3N 暴露于所需的双键。然后用Burgess试剂脱水得2。
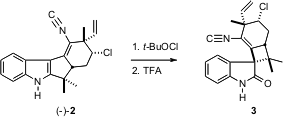
利用通过制备1建立的系列的绝对构型,生物碱2的对映体由S-卡酮制备。两个- [R -香芹酮和小号 -香芹酮是廉价的和可用散装。将如此制备的2暴露于t- BuOCl,接着三氟乙酸诱导氧化重排成(+) - 韦特吲哚A(3),与天然产物相同,包括旋转符号。
这三种合成方法显着地进行而不依赖于官能团保护,强调了由7和8的联合所示的氧化吲哚偶合的能力,得到9。作者认为,与试图从11制备2时遇到的困难相比,12到2的容易转化可以表明它实际上是轴向胺12或作为2的生物合成前体的衍生物。


目前评论: