- A+
标题:使用亲核有机催化剂从烷基亲电子试剂中光化学生成自由基
作者:Bertrand Schweitzer-Chaput, Matthew A. Horwitz, Eduardo de Pedro Beatoand Paolo Melchiorre*
(Nat. Chem. 2019, 11, 129−135)
报告人:屈中华
导语:
传统上,产生自由基需要利用键离解能或前体的氧化还原性质的策略。近日,Paolo Melchiorre课题组公开了一种光化学催化方法,其利用底物的不同物理性质以形成碳自由基。他们使用亲核二硫代氨基甲酸盐阴离子作为催化剂,用精心定制的发色单元装饰,通过SN2途径活化烷基亲电子试剂。所得光子吸收中间体在由可见光诱导的均裂裂解时提供自由基。
主要内容:
确定有效的自由基生成策略可以促进该领域的发展。一种传统方法依赖于引发剂。引发剂是由热或高能光诱导的均裂时产生可从底物中提取氢或卤素原子的反应性基团(图1a,左图)的高能化合物(例如重氮化合物或过氧化物)。该方法最终依赖于底物的键离解能(BDE)以形成目标开壳中间体I。另一种通向自由基的经典途径利用了可以通过化学计量的氧化剂/还原剂或通过电化学方法触发(图1a,右图)底物参与氧化还原过程的趋势。然后,从单电子转移(SET)结果中出现的自由基离子可以碎裂以产生目标基团I。这些经典策略很有用。然而,它们需要危险和有毒试剂,高温和/或UV光照射等相对苛刻的条件,总体上限制了随后的自由基过程的选择性和官能团耐受性。
朝着更温和的反应条件以及更具选择性的反应迈进的关键步骤,是使用具有硫代官能团并充当自由基前体和反应物的底物(图1b,左)。由Barton引入(Barton, D. H. R. & McCombie, S. W. A new method for thedeoxygenation of secondary alcohols. J. Chem. Soc. Perkin Trans. 1975, 1, 1574–1585)并由Zard进一步推广的黄原酸盐转移化学方法(Zard, S. Z. On the trail of xanthates: some new chemistry from an old functional group. Angew. Chem. Int. Ed. 1997, 36, 672–685)极大地扩展了自由基的合成潜力,但仍依赖于特意设计的化学计量试剂。光氧还原催化提供了一种在非常温和的条件下以催化方式获得自由基的有吸引力的方法。这种方法利用了光吸收金属或有机催化剂的以利用光子能量重复地从除去电子或捐赠的电子到简单台式稳定的底物(图1b中,右)的能力。这种SET机制通常用于生产目标自由基I,但是一些光氧还原催化剂也可以采用氢提取歧管来生成I.与所有其他自由基生成策略一样,当应用光氧化还原催化时,化学家必须依赖于氧化还原性质或BDE预测给定底物是否适合成功形成目标开壳物种I。
在这里,作者报告了一种利用底物的不同物理性质来产生碳自由基(图1c)光化学催化策略。具体而言,作者表明,用合适的发色团装饰的1型亲核二硫代羰基阴离子催化剂可以经历具有不同离去基团的简单烷基亲电子试剂的SN2置换。得到的光吸收中间体II在弱可见光(蓝色发光二极管,LED)激发和弱C-S键的均裂解裂时提供自由基。作者使用这种基于SN2的歧管从各种对于经典的自由基生成策略是惰性的或不相容的底物产生开壳中间体。该方法的温和反应条件和高官能团耐受性允许C-C键形成反应的发展,市售药物的简化制备,活性药物成分和生物相关化合物的后期衍生化,以及对映选择性自由基催化。

设计方案:图2a详述了作者提出的通过利用前体的亲电特性产生自由基的策略。作者设想了催化循环,其中二硫代羰基阴离子1将用作在SN2进攻和离去基团(LG)的置换时活化烷基亲电试剂2的亲核催化剂。得到的中间体II具有可被低能光子裂解而产生目标碳基自由基III和二硫代羰基自由基IV弱的C-S键。然后,开环中间体III将被缺电子的烯烃3拦截,以提供新的C-C键。新出现的亲电子基团V反过来从1,4-环己二烯(1,4-CHD)中提取氢原子,从而形成最终产物4和环己二烯基VI。来自环己二烯基VI的二硫代羰基IV的放能SET还原,最终将通过返回催化剂1来关闭催化循环。
方法优化:为了验证作者的推测,作者研究了富马酸二甲酯3a的Giese型自由基共轭物(图2b)。使用市售的γ-萜品烯作为1,4-CHD的更便宜且更稳定的替代物,在乙腈(CH3CN)中进行实验。当使用乙基黄原酸钾1a作为亲核催化剂(10mol%)并在400nm照射下进行模型反应时,以19%的收率得到所需产物4a(条目1,图2b)。该初始结果表明催化产生苄基的可行性。通过将光源改变为在465 nm发射的蓝色LED完全抑制反应(条目2)。作者从SN2过程中出现的黄原酸酯中间体IIa不能吸收蓝光(图2c)的这种反应性行为是合理的。亲核催化剂1的模块化性质允许作者包括发色团单元,一种旨在增强II型关键苄基中间体吸收性能的结构修饰。分别带有咔唑和吲哚支架的硫代氨基甲酸酯催化剂1b-c在与底物2a进行SN2反应时提供中间体IIb-c。这些中间体相对于IIa具有显着增加的摩尔消光系数。那些改进的光学性质转化为在蓝光照射下恢复的催化活性(λmax= 465 nm,条目3和4)。当使用含吲哚的硫代氨基甲酸酯催化剂1c时,将温度升至60℃使产物3的产率达90%(条目5)。在控制实验下,在没有催化剂1或光的情况下没有检测到产物形成。在自由基抑制剂2,2,6,6-四甲基哌啶1-氧基(TEMPO,1当量)的存在下也观察到反应性的抑制,而使用非脱气条件只略微降低了产率(条目6)。从机理上讲,作者还评估了在反应过程中产生VII型(如图2a所示)的形式上的二硫代氨基甲酸酯基转移产物的可能性。尽管作者从未在催化条件下检测到该加合物,但是当用蓝色LED照射并且存在γ-萜品烯时,VII型中间体的真实样品提供产物4a。该观察结果与二硫代羰基化合物向基团转移歧管的趋势一致,表明加合物VII可以是与祖先自由基IV和V平衡的光活性物质。
为了证明该方法的稳定性和对氧的相对耐受性,该反应可以在不对溶剂进行脱气的简单的试管中有效地进行,并且使用水作为共溶剂(83%产率,条目7)。该方法操作简单,结合使用廉价试剂和空气和水分稳定的固体催化剂,使该反应非常适用于使用常用玻璃器皿和光照射设备的多克规模应用(73%产率,8.6 g)。值得注意的是,可以通过考虑不同底物的溶解度来选择溶剂,当在各种反应介质中(例如乙酸乙酯,二氯乙烷,甲苯和四氢呋喃均提供85-90%的产率)进行模型反应时没有观察到反应性的显着差异。
该策略的机理表明,具有适合SN2置换的离去基团的其他苄基底物可用于模型反应。因此,带有碘化物,溴化物,甲磺酸盐或三氟乙酸盐的不同前体2b-e以高产率提供产物4a(图2d)。苄胺衍生的Katritzky盐2f也是合适的底物。因此,该活化模式不限于卤化物,也可以使用胺和醇衍生物。相反,具有不良SN2过程倾向的离去基团,包括乙酸盐和磷酸盐,完全未反应。因此,一个离去基团或另一个基团的选择可以由其易于进攻或与复杂合成计划中的其他官能团的兼容性决定。

自由基生成策略:作者在不同的C-C键形成转化的背景下评估了这种光化学自由基生成策略的合成潜力(图3)。作者首先探讨了由亲核催化剂1c催化的富马酸盐加成的Giese型的范围(图3a)。具有富电子和贫电子芳基取代基(2g-j)以及二级苄基底物(2k)和氯化丙烯基2l(烯丙基自由基前体)多种苄型亲电子试剂均为有效底物,以中等产量提供相应的共轭加成产物。该策略可用于从恶唑烷酮,受保护的仲胺或受保护的伯胺中引入α-氨基甲基化方法的底物2m-o。然后,作者评估了与未保护的极性官能团的相容性2t-2w,均在所需的苄基位置被耐受和活化。最后,作者证明了市售N-(氯甲基)邻苯二甲酰亚胺2o的Giese型反应可以与各种迈克尔受体一起反应(图3b)。带有各种吸电子基团的烯烃(例如酯,腈,砜和酰亚胺)都以良好的收率反应,产生加合物5a-e。
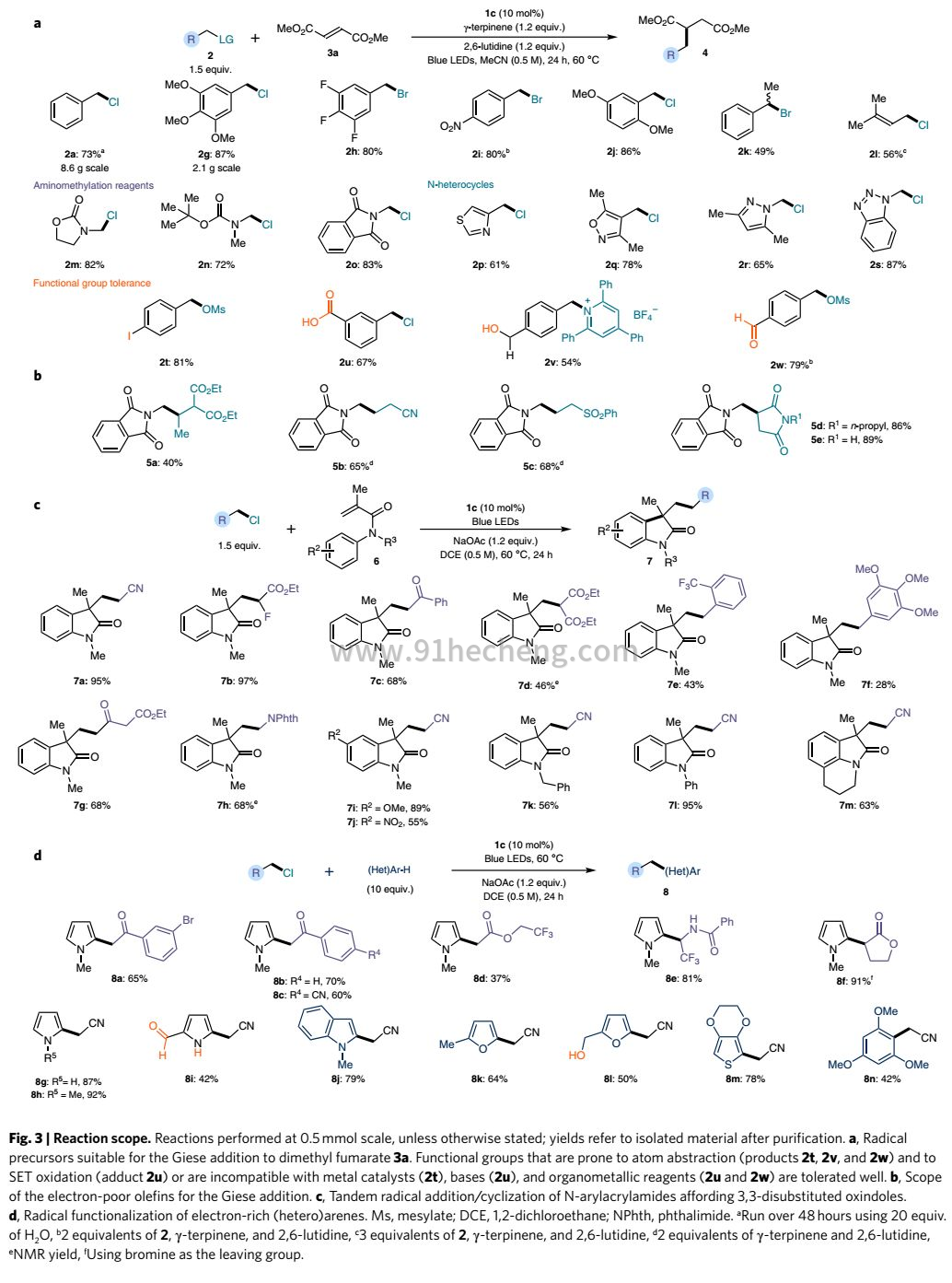
接着,作者应用作者的催化体系开发其他合成有用的自由基C-C键形成过程。根据作者的机理推测,作者推断VI型的环己二烯基是一种作为催化剂转化的还原剂(图2a)关键的中间体,可以通过富电子芳香体系自由基捕获产生。避免使用化学计量的H原子供体的这种机理变化被成功地实施以使得能够制备官能化(杂)芳族产物。由催化剂1c促进的芳族丙烯酰胺6的顺序自由基加成/环化提供了一系列不同取代的氧化吲哚7(图3c)。类似地,富含电子的(杂)芳族化合物(包括未保护的吡咯(8i))的直接自由基官能化是从容易获得的烷基氯作为自由基前体开始进行的(图3d)。许多如醛,仲酰胺和游离醇等反应性官能团,导致合成有价的取代(杂)芳族化合物8。
总结:
总之,Paolo Melchiorre课题组报告了一种利用从与传统的生成自由基的策略不相容或不活泼的各种亲电底物中生成开壳中间体的SN2过程(离子化学的基本途径)的光化学策略。该方法需要易于获得的有机催化剂并且在可见光照射下发生。底物范围研究表明,未保护的极性官能团和N-杂环都是良好耐受的。初步结果强调,这种自由基生成策略可用于包括光驱对映选择性催化更复杂的环境。这些发现以及实验简单性和催化剂的低成本表明该方法将发现进一步的合成应用并且可能适用于药物化学和工艺开发。


目前评论: