- A+
Helicterin B,Helisorin和Helisterculin A的全合成
斯奈德
SA Snyder,F。Kontes,J。Am。化学。SOC。 2009年,131,1745年至1752年。
DOI: 10.1021 / ja806865u
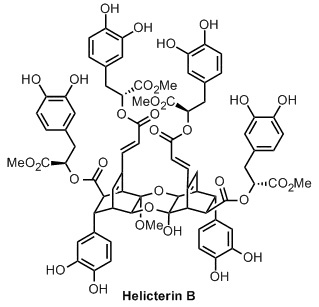
Helicterin B和相关化合物是二聚(和四聚)天然产物,可以使用retro-Diels-Alder / Diels-Alder级联来构建。Porco完全合成了 chamaecypanone C和Synder合成的helicterin B,通过复古Diels-Alder / Diels-Alder级联的关键用途联合起来,两者都引用了Bedekar早在1992年的一篇明显鼓舞人心的论文 。
那么除了它的大小(1510 g mol -1)之外,这个非常大的分子有什么新东西?并不多,为了公平 - 斯奈德提到了“对禽类成髓细胞瘤病毒的轻度抑制活性”,但当作者将这种活动描述为温和时,你可以相当肯定它几乎不活跃。因此,我们必须明确关注天然化合物的构建。
Synder从稍微简单的helisorin的合成开始。合成开始于迷迭香酸衍生物的二聚化,其分五步产生,然后用对 -三氟甲基苄基选择性保护 。经过一些实验后,这个充满异国情调的保护组被使用,Snyder试图在最后一步找到合适的组。由于目标的功能,这意味着去除不依赖酸,碱或氧化脱保护 - 限制他们的选择。所以他们发现了这种'OTfBn醚',它可以使用中等强度的路易斯酸去除,但是温和稳定。
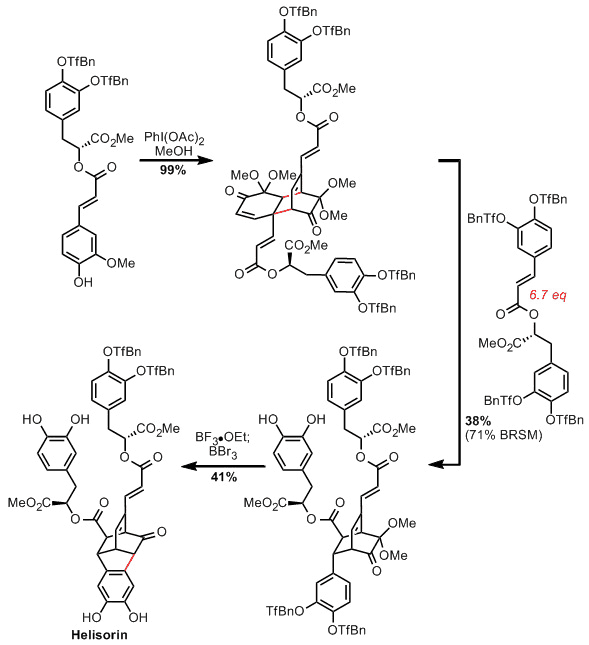
然后使用高价碘试剂(PIDA)将迷迭香酸衍生物氧化二聚化。然后用亲二烯体(也由迷迭香酸产生)加热该中间体,诱导逆Diels-Alder / Diels-Alder级联并产生Helisorin的核心。然而,仍然需要形成另外一个碳 - 碳键,并且它们的保护基团选择变得清晰 - 使用温和的路易斯酸(LA)使近端保护的儿茶酚环化成双环辛烷,而更强的LA将保护基团移除到完成这个目标。问题是,为什么较强的洛杉矶不用于这两个步骤:好吧,显然效果不好,导致二甲基缩酮破裂。
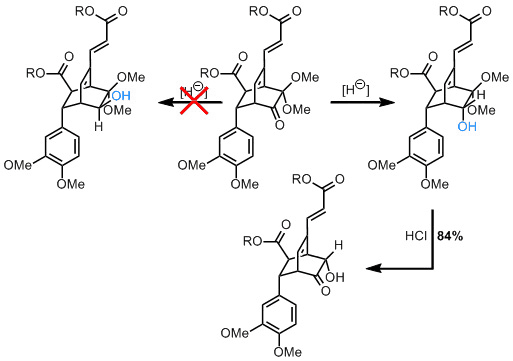
随着这一成功,该小组已准备好转向更大的目标,但他们对螺旋体双环核心的微妙不同的立体化学存在问题。在Helisorin合成中转化倒数第二个中间体是关键,但需要从不太有利的方法中减少酮。使用基于氢化物的途径是无效的,但是他们能够通过进行“平衡的酮醇转换”来使用不需要的产物。该反应的产物,即第二种酮,更容易接受还原,可能是通过从所需面上配位还原剂(Me 4 NBH(OAc)3)。有点幸运,但干得好。
更进一步,他们有双环辛烷类化合物恰到好处。较弱的LA引起该中间体的二聚化,完成了大部分天然产物。然而,p- CF 3 - 苄基醚的最终脱保护如前所述不太成功,因为甲基也在反应中损失。这意味着他们没有完成螺旋体A,但幸运地发现他们已经分离出了螺旋体B!
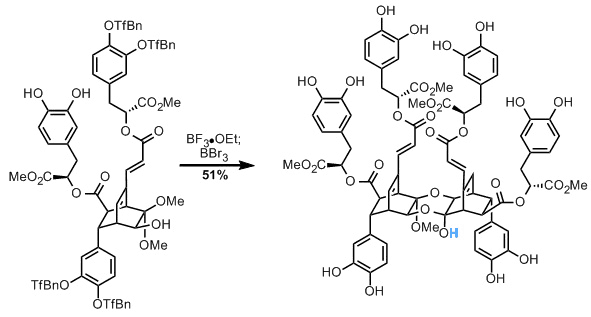
使用相同的常用中间体可以轻松完成helisterculin A!这是一项伟大的工作,并且总是很高兴在一篇适当的全文中阅读它。


目前评论: