- A+
膦催化[4 + 2]环化:6-苯基-1-甲苯磺酰基-1,2,5,6-四氢吡啶-3-羧酸乙酯的合成
讨论
已经有显着的进步在有机膦催化的反应,尤其是涉及处理[4C + X] annulations,因为它们在构建5-8元的环状产物的潜在应用。我们小组在2003年首次报道了2-烷基 - 丁-2,3-二烯酸酯和醛亚胺之间的膦催化的[4 + 2]环化,已经成为构建取代的四氢吡啶衍生物的有力工具(方案1)。3根据普遍接受的机制,亲核加成三- Ñ丁基膦以在形成共振的α -烷基的联烯酸酯的结果β位稳定的两性离子物种阿。亲核加成烯醇化物的阿成Ñ-tosylimine 2产生磺酰胺乙。通过质子转移,物质B与乙烯基鏻叶立德C / D平衡 。再一次质子转移促进E中磺酰胺阴离子的形成,其经历共轭加成到α,β-烯酸酯,然后β-脱去三丁基膦,导致四氢吡啶3的形成。本讨论附录侧重于此[4 + 2]环形的新发展 - 包括其不对称版本和合成效用 - 自2003年以来。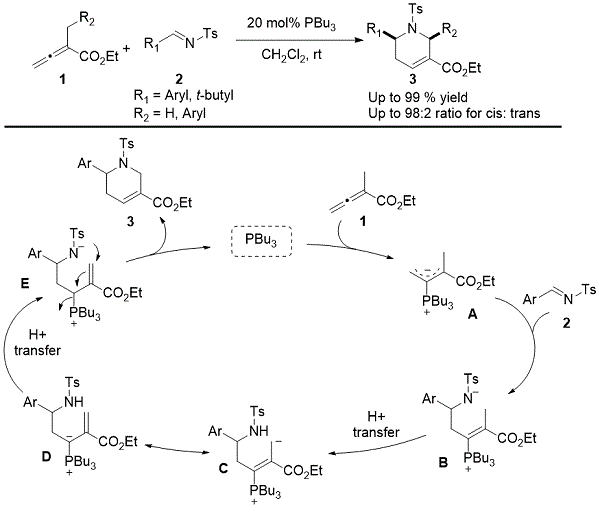
方案1.由Kwon制备α-烷基芳酸 - 亚胺[4 + 2]环的机理
当在三丁基膦存在下使2-取代的2,3-丁二烯酸酯和N-甲苯磺酰亚胺反应时,Kwon的[4 + 2]环化进行以提供具有高效率和非对映选择性的四氢吡啶。具体地,α-甲基月桂酸乙酯与各种N-甲基芳基亚胺进行[4 + 2]环化以提供四氢吡啶,通常以超过90%的分离产率。丙二烯 - 亚胺[4 + 2]环化是一个稳健的过程,已经报道了四氢吡啶的克规模制备。3
虽然2-甲基-2,3-丁二烯酸酯与N-甲苯磺酰亚胺经历有效的[4 + 2]环化,但3-甲基-3,4-戊二烯酮经历了令人惊讶的级联事件,在该过程中引入了两个亚胺分子。例如,在2005年,Shi提出了在三丁基膦存在下由3-甲基-3,4-戊二烯酮和N-甲苯磺酰亚胺合成肉桂基四氢吡啶酮的罕见实例(方案2)。4该反应生成的烯醇化物鏻ģ作为中间负责将亚胺的另一单元和得到相应的四氢吡啶基肉桂酮。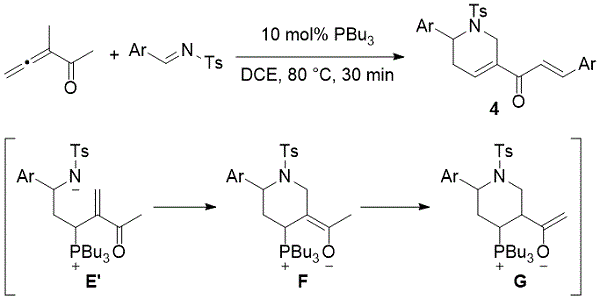
方案2.施氏四氢吡啶衍生物的形成
2012年晚些时候,Ye和同事在[4 + 2]环中使用糖精衍生的环状酮亚胺,可以快速获得各种功能化的三环四氢吡啶(方案3)。五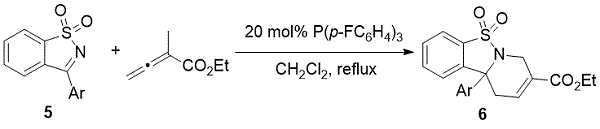
方案3.Ye的官能化多环四氢吡啶的合成
2014年,郭及其同事发现环状氨基磺酸盐是膦催化[4 + 2]环化方案的亚胺组分(方案4)。6将反应物在制备氨基磺酸三环高效8以高收率。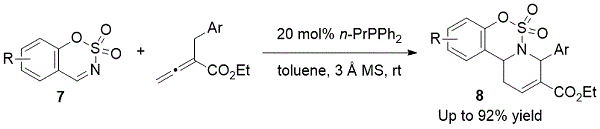
方案4. Guo的官能化四氢吡啶的制备
对映选择性亚乙基亚胺[4 + 2]环化:已经报道了一些不对称形式的Kwon的丙二烯 - 亚胺[4 + 2]环化。2005年,Fu展示了不对称[4 + 2]反应的优雅实例,当使用Gladiali的磷脂酰(R)-P1作为催化剂时,产生官能化的四氢吡啶(方案5)。7反应以几乎定量的产率提供四取代的四氢吡啶,具有优异的非对映选择性和对映选择性。使用亚乙烯基琥珀酸盐,即α-乙氧基羰基甲基月桂酸酯,以确保高反应性和选择性。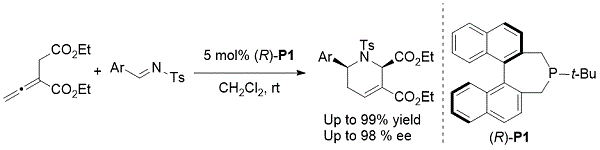
方案5. Fu的不对称丙二烯 - 亚胺[4 + 2]环
2011年,Zhao引入了氨基酸衍生的双功能N-酰基氨基膦催化剂P2,用于不对称丙二烯 - 亚胺[4 + 2]环化。8此反应,得到手性系列四氢吡啶衍生物的具有高对映选择性优异的产率(方案6)。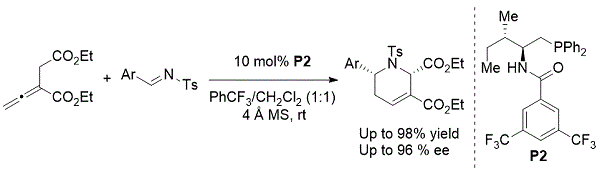
方案6. Zhao的对映选择性制备四氢吡啶
Sasai报道了2014年从环磺酰亚胺中对映选择性合成多环四氢吡啶(方案7)。9手性螺 - 磷杂环庚烷催化剂(R)-SITCP(P3)促进了糖精衍生的酮亚胺和α-甲基月桂酸乙酯的对映选择性形式[4 + 2]环加成反应。这些反应以良好的产率和对映体过量提供了三环四氢吡啶,具有优异的区域选择性。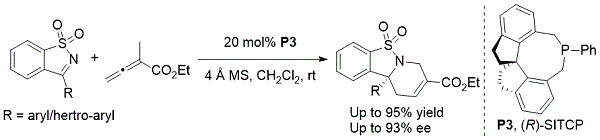
方案7. Sasai不对称合成官能化三环四氢吡啶
同年,郭和同事证明,当使用基于氨基酸的双官能膦P4作为手性催化剂时,形成了对映体富集的环状氨基磺酸酯(方案8)。10这种不对称[4 + 2]环加成反应提供了高产率的手性氨基磺酸盐稠合四氢吡啶,具有优异的对映选择性。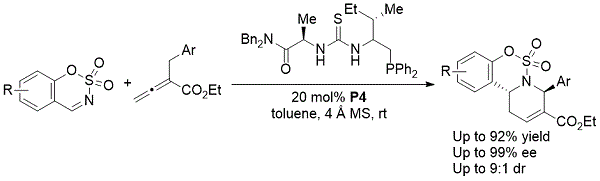
方案8. Guo的环状氨基磺酸酯的不对称合成
最近,在2018年,我们的小组报道了通过[4 + 2]亚胺与α-甲基月桂酸酯的环化催化对映选择性合成瓜氨酸衍生物。11甲P-手性[2.2.1]双环膦,外切 - (p -anisyl)-HypPhos (P5),在α-alkylallenoates和亚胺之间的反应施加,产生对映体富集guvacine衍生物(方案9)。该方法用于通过高产Tebbe烯化/水解序列合成对映体纯的aplexone 10。对斑马鱼胚胎的aplexones的表型分析显示,(R) - 哌啶酮是斑马鱼胚胎中胆固醇细胞水平降低的原因。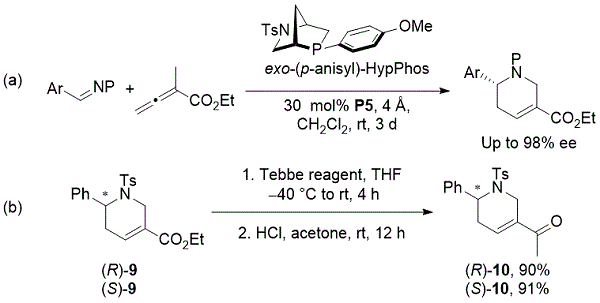
方案9. Kwon的催化对映选择性合成guvacine衍生物
[4 + 2]环化反应的应用:在天然产物的全合成中丙二烯 - 亚胺[4 + 2]环化反应的潜力首先在(±) - 醛固酮的正式合成中示出(方案10)。12二乙基2- vinylidenesuccinate和吲哚亚胺的处理11与催化PBU 3得到indolytetrahydropyridine 12以良好的收率以3:1博士。然后将四氢吡啶12转化为已知的中间体13通过一系列六步转化获得优异的产率,包括Friedel-Crafts酰化,nosyl基团脱保护,胺的甲基化,还原脱氧,乙酯的选择性1,2-还原。库克和同事之前曾报道过烯丙醇13合成了( - ) - 羊甾氨酸(14)。13,14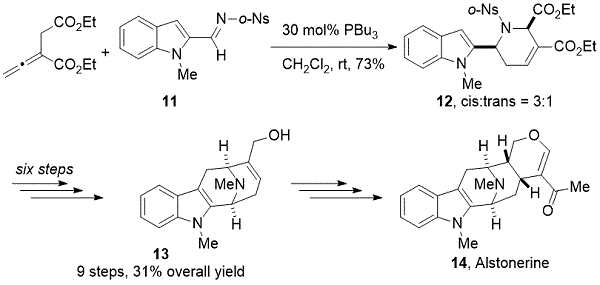
方案10. Kwon正式全合成(±)-alstonerine
2012年,我们小组对(±)-hirsutine的全合成应用了类似的[4 + 2]α-亚甲基乙酸乙酯与亚胺的环化(方案11)。15膦催化[4 + 2]粗N-(邻 -甲苯基)亚胺与α-甲基月桂酸乙酯的环化,得到四氢吡啶,两步产率为73%。由粗亚胺以良好收率形成四氢吡啶显示出该反应的稳健性。随后的官能团操作导致C-环的构建并且以高效率完成(±) - 多萜烯的全合成。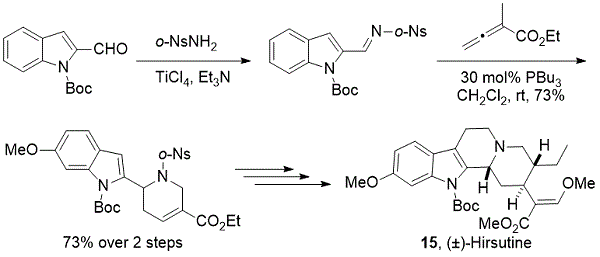
方案11. Kwon全部合成(±)-hirsutine
2012年晚些时候,我们的研究小组对利血平的五环骨架进行了简明的制备(方案12)。16之间的[4 + 2]环ñ - (ö -nosyl)亚胺和乙基α-methylallenoate在催化PBU的存在3提供,以良好的收率,具有A-,B-,和d-环的四氢吡啶中间体利血平。从关键中间体以两步开始进一步构建C-环,随后进行6π-电环化以形成E-环,提供利血平的五环支架16。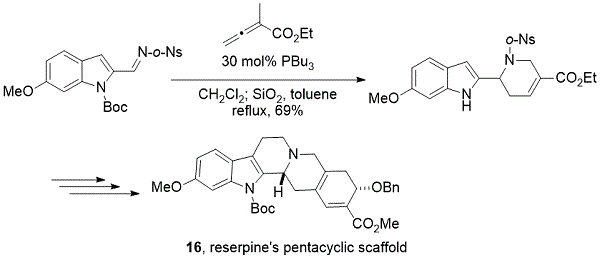
方案12. Kwon进入利血平的骨骼框架
2007年,我们的研究小组报告了使用聚苯乙烯结合的烯醇化物制备组合文库的磷化氢催化的第一个例子,以及鉴定蛋白质香叶基香叶基转移酶I型(GGTase-I)和Rab香叶基香叶基转移酶作为潜在的抗癌治疗剂的有效抑制剂。17,18,19
使用用Wang树脂接枝的SynPhase灯,将烯醇酸与Wang接头的苄醇单元偶联以安装树脂结合的烯醇化物17(方案13)。通过分裂和池策略,结合了大量的烯醇酸,亚胺和硫醇结构单元,导致4288种化合物的生产,包括各种各样的官能化四氢吡啶,吡咯啉,吡咯烷和哌啶。体外测定显示吡咯啉 18和四氢吡啶19均显示出针对GGTase-1的亚微摩尔IC 50值。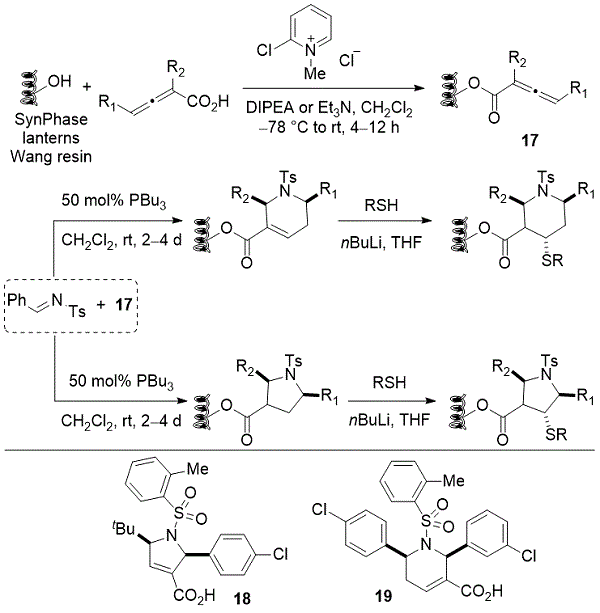
方案13. Kwon的膦催化合成GGTase-1抑制剂文库
吡咯啉18的衍生物在小鼠中显示出针对固体胰腺肿瘤和肺癌模型的体内功效,暗示了开发新型抗癌治疗剂的可能性。20,21
亚胺和烯醇化物的膦催化产生具有α,β-不饱和羧酸酯官能团的吡咯啉和四氢吡啶,其可以转化为多环支架。2011年,我们的小组披露了一种化学文库的多样性导向合成,该文库包含91种具有16种不同支架的多杂环化合物(方案14)。22,23含有吡咯啉和四氢吡啶中的α,β-enoate单元是通过将Tebbe烯转化成相应的二烯醇醚,以及随后进行与各种亲二烯体的Diels-Alder反应,从而提供密集的官能多杂环化合物。兴高采烈,化合物20 - 22 显示出针对MDA-MB-231人乳腺癌细胞的亚毒性抗迁移活性。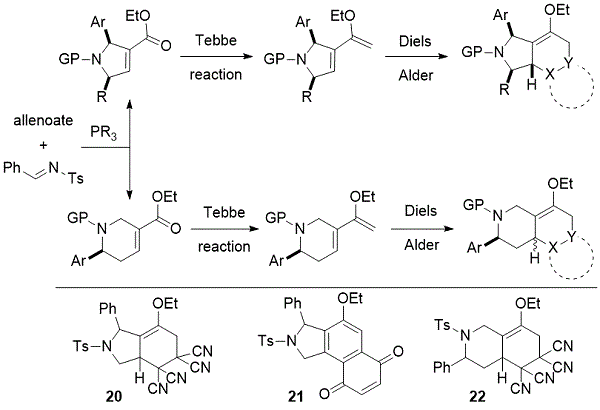
方案14. Kwon以多样化为导向的多杂环化合物库的合成
利用Wang树脂结合的四氢吡啶的容易制备,通过在固相上的分裂池合成来构建八氢-1,6-萘啶-4-酮的文库。24,25同样,联烯酸被耦合到所述Wang树脂,接着[4 + 2]与三丁基膦的存在亚胺annulations(方案15)。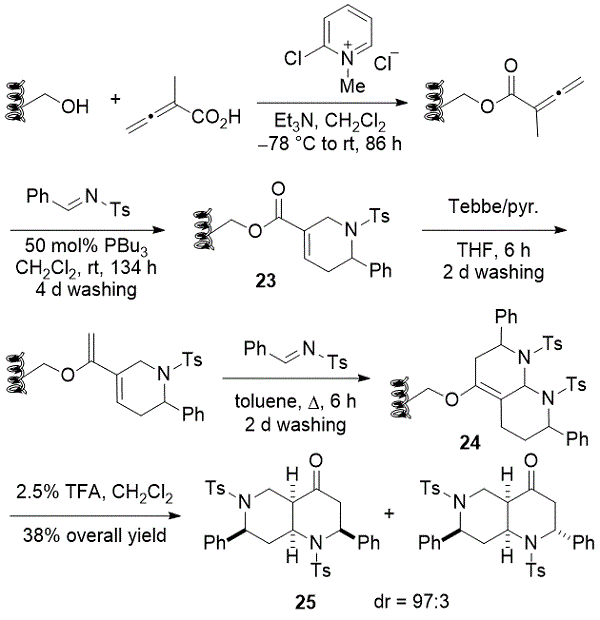
方案15. Kwon形成八氢-1,6-二氮杂萘-4-酮
用Tebbe试剂处理 所得的四氢吡啶羧酸酯23,然后用亚胺进行Diels-Alder反应。值得注意的是,相同的亚胺结构单元用于膦催化和Diels-Alder反应。八氢萘啶24的高度非对映选择性水解在用三氟乙酸(TFA)简单处理后发生,释放出萘啶酮25。间的96个naphthyridinones,五独特的八氢-1,6-二氮杂萘-4-酮26 - 30显示内皮细胞的活化优异的触发先天性免疫应答的诱导(方案16)。这些研究说明了膦催化产物的潜在用途。从溶液到固相的原始膦催化反应的现成翻译使得通过分裂 - 池组合合成 - 现代化学生物学的关键方面 - 容易地制备类似物。
方案16. Kwon的免疫活化八氢-1,6-萘啶-4-酮
近年来已经实现了增强[4 + 2]环的效用的若干进步。已证明这些反应在许多不同的环境中通常是有效的,包括固相和手性膦的影响。因为四氢吡啶核心出现在无数的天然产物和生物活性分子中,我们预计丙二烯和亚胺之间的这些[4 + 2]环将继续用于有价值的产品合成,特别是那些温和条件,强效和高选择性是至关重要的。

我的微信
关注我了解更多内容

目前评论: