- A+
虽然发现P(CH3NCH2CH2)3N(1)不如1,5-二氮杂双环[4.3.0]非5-烯(DBN)或1,8-二氮杂双环[5.4.0]十一碳-7-烯有效(DBU)从维生素A中间体13-顺-10-溴-9,10-二氢精氨酸乙酯(6)和14-溴-9,14-二氢化合物乙酸酯(11)中除去溴化氢时,反应在回流苯,在室温下在乙腈中,它优于DBN和DBU。该反应的31P NMR研究表明,在1的存在下由乙腈-d3产生的碳负离子是引发消除步骤的基本物质。(Z)-HC≡C(CH3)=CHCH2OH亲核加成到(E)-2-甲基-4-(2’,6’,6’ - 三甲基-1-环己烯-1¢的羰基)的非对映选择性 - ) - 3丁烯醛(2)仅为中等(20%),并且(9R *,10S *) - 13-顺式11,12-二脱氢-9,10-二氢-10-羟基 - 视黄醇(3b)占优势。非对映异构二醇3中C≡C键的LiAlH 4还原得到13-顺式-9,10-二氢-10-羟基乙烯醇4a和4b作为主要产物以及11个顺式-13-顺式异构体和脱氧化合物(3EZ,5EZ, 8E)-3,7-二甲基-9-(2,6,6-三甲基-1-环己烯-1-基)-1,3,5,8-壬烯(9)。纯的非对映异构烯丙基醇4a和4b的15个乙酸酯与PBr3的反应发生,具有显着但不相同的构型保留,并伴随形成重排的溴化物11。
介绍
在该实验室中化合物1的合成以及其非常强碱性的发现促使我们评估1在从有机底物中消除H-X物质(X)Cl,Br,I,OSO3R等中的反应性。作为该项目的一部分,我们将注意力集中在通过将第五个CdC键引入视黄醇骨架的9,10位来合成全反式维生素A及其各种异构体的方法。尽管使用DBN成功地完成了全反式维生素A的合成,但由于后一化合物的较高碱性,可以设想通过使用1改善该合成。在这里,我们报告了这种改进,并提出了1的行动模式的机制。

结果和讨论
合成。通过消除溴化氢合成维生素A的主要步骤在方案1中描述,并与文献中描述的那些相同。然而,优化序列的每个步骤,并分离和鉴定所有转化的主要副产物。通过在相转移催化(PTC)条件下从三甲基锍甲基硫酸盐引入碳原子,然后根据文献程序将中间环氧化物转化为2,以98%的总收率,从β-紫罗兰酮获得所需的醛2。试图通过Darzens反应从β-紫罗兰酮合成纯2是不成功的,导致含有2的混合物被其R,α-异构体和其他可能具有聚合性质的杂质污染。
方案一

试剂:(i)HCtC(CH3)CdCHCH2OH,2BuLi,THF水; (ii)醚中的LiAlH4; (iii)2,4,6-可力丁中的Ac2O; (iv)PBr3在乙醚中,-20°到rt; (v)1,DBN或DBU
通过C14 + C6偶联合成视黄醇骨架通常是通过将2-甲基-2-戊烯-4-基-1-醇的二镁盐加入醛2或优先加入其R,β-异构体来实现的。。尽管反应缓慢,但可以使用锂盐加速反应。因此,顺式-2-甲基-2-戊烯-4-基-1-醇的二锂盐(通过向醇的四氢呋喃(THF)溶液中加入2当量的丁基锂得到)用2处理,得到 13-顺式-11,12-二脱氢-9,10-二氢-10-羟基苯乙醇(9R *,10R *) - 3a和(9R *,10S *) - 3b的2:3混合物,分别为85 然而,发现粗产物的收率为%,足以用于随后的转化。由1H NMR光谱数据确定的非对映异构体的比例与反应条件以及阳离子(即Li +或Mg2 +)无关。
炔键与LiAlH 4的反应通常产生纯的反式产物,而当在失活的钯催化剂上进行氢化时,预期独特地形成顺式异构体。在我们的手中,LiAlH 4还原为3,得到所需的非对映异构体11,12-反式二醇4和11,12-顺式二醇8的混合物,其比例为约4:1,总产率为65%。通过硅胶色谱法容易地分离该混合物。此外,还获得了非极性级分(5%),后来被鉴定为四种异构烃的液体混合物9。通过柱色谱法实现非对映异构二醇4的部分分离,之后使用极性更大的二醇4b(洗脱)第二列来自结晶。粗还原产物中(9R *,10R *) - 4a与(9R *,10S *) - 4b的比率反映了非对映异构体化合物3的比例为2:3。对于NMR表征,非对映异构体顺式二醇8被分离。经过几次色谱分析后在硅胶上进行。

使用2,4,6-可力丁中过量的乙酸酐,以可接受的选择性实现4a和4b中伯羟基的选择性保护作为相应的乙酸酯。当使用2当量的酸酐和过量的碱时,完成将纯4b定量转化为单乙酸酯(9R *,10S *) - 5b和二乙酸酯10b的9:1混合物。使用氯仿或二氯甲烷作为溶剂并且吡啶作为碱仅导致起始二醇4的不完全转化和过量形成二乙酸酯。当乙酰氯用作乙酰化试剂时,形成甚至更复杂的混合物。
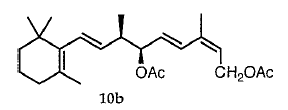
用三溴化磷最可令人满意地用二级烯丙醇5合成非对映异构体溴化物6。其他溴化试剂,即PPh3 / NBS和PPh3 / CBr4,在复杂反应混合物中仅产生痕量的6。用PBr3将羟基转化为溴化物也提供了有趣的区域和立体化学结果。与(9R *,10R *) - 5a与(9R *,10S *) - 5b的比例无关,形成约10-30%的重排溴化物11。此外,当用PBr3处理纯5a时,几乎完全转化为含有大约5的(9R *,10R *) - 6a和(9R *,10S *) - 6b(3:1)的混合物。观察到11%的10%。在相同条件下,纯5b得到6a和6b(3:7)和23%的11的混合物。
最初,我们研究了在回流的苯中将6和11的混合物转化为异构的乙烯基视黄酯9-顺-13-顺-7a和9-反-13-顺-7b(表1)。发现在等摩尔量的1,5-二氮杂双环[4.3.0]壬-5-烯(DBN)或1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)存在下,非对映异构体6a和6b的混合物在15分钟后几乎定量转化为7a和7b的混合物,而重排的溴化物11仅部分地消除。在回流的甲苯中用DBN进行的相同转化在15分钟后仅留下痕量的未反应的11。
通过1H NMR光谱监测在室温下从6a,6b和11的混合物中用1,DBN或DBU在乙腈中消除溴化氢的进展。发现1比DBN或DBU更具反应性,导致在30分钟内6和11的信号消失,而仅在1小时后观察到完全转化为DBN。在这些条件下使用DBU,1小时后仍然残留未反应的6和11的痕量。

脱卤化氢反应产物的纯化包括水处理和通过失活氧化铝垫过滤,得到由7a和7b组成的异构视黄基乙酸酯的混合物作为主要产物(参见表1)。从6a和6b中消除溴化氢仅导致13-顺式异构体7a和7b。另一方面,从重排的溴化物11中,发现全反式 - 视黄基乙酸酯(12a)及其9-顺式异构体12b的形成如预期的那样,而不是更具空间应变的13-顺式异构体7a和7b。当溴化物6和11在乙腈中用1在30分钟内进行脱溴化氢时,注意到维生素A异构体的产率没有提高。来自DBU,DBN和1-诱导的脱氢溴化的粗产物的1H NMR光谱的比较表明,乙腈中的1可以诱导高度共轭产物的一些分解。当溴化物6和11的混合物用1℃的乙腈溶液在0℃处理2 1/4小时时,观察到不完全转化。
表1.溴化物6和11中HBr消除的产率和立体化学结果
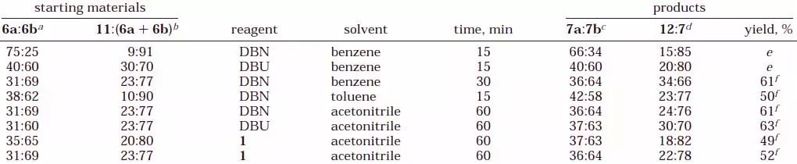
a基于H12信号的相对强度。 b基于H9信号的积分。 c基于H3C18信号的积分。 d基于H14信号的积分。 e反应进行到95%以上完成。 f对于仅含有7a,7b和12的混合物。
结构,立体化学和机械方面的考虑因素。最初,我们对使用1的脱溴化氢转化率低于DBN或DBU在回流苯中的转化率感到失望。事后看来,与DBN和DBU相比,这一观察结果可以合理地确定1中碱性磷中心周围较大的空间体积,特别是当它变为质子化并转化为三角双锥体阳离子时,正电荷在1上形成。在乙腈中然而,1比DBN和DBU在室温下从6和11中消除HBr的优越性源于1的碱度(在乙腈2b中pKa为13 = 33)与DBU的碱度(pKa = 23.9)和DBN(pKa = 23.4)。尽管13的pKa与乙腈的pKauto 33的报告值相同,但已证明该自动溶解常数的值为44。在任何情况下,我们发现非离子碱如1能够以足够的效率将乙腈去质子化为-CH2CN,使其在影响伯,仲和叔烷基卤化物的脱卤化氢作用方面起到空间小但有效的作用。亲核试剂从与醛和酮的反应合成3-羟基腈
在目前的工作中,-CH2CN的生成得到了31P NMR研究的支持,该研究从6和11中消除了溴化氢,其中1为乙腈3。在加入溴化物之前,1的氘代乙腈溶液的31P NMR光谱显示1(单线态在120.8ppm)和微量的氘代阳离子(三条相等强度的线在-10.0ppm)的信号。当脱溴化氢反应完成时,仍然存在少量过量的1,而超过80%的1转化为其氘代形式,并且发现少于20%作为质子化物质。
7b和9-反式7b-顺式构型的证据由它们的1H NMR光谱提供。虽然发现两种异构体中H7-H8和H10-H11-H12质子组之间的类似偶合常数之间没有差异,但注意到7a中的H8相对于7b中的H8被去除了0.5ppm。当全反式和13-顺式 - 视黄醛和甲基维甲酸中的H8的化学位移分别与9-顺式 - 和9-顺式-13-顺式异构体进行比较时,发现了相同的现象。全反式12a和可能还有9-顺式-12b的存在是从观察到Hä3.58ppm处的H14三联体和该质子与其在1H-1H COSY谱中的1.86ppm处的H3C2的长程相关性得出的。已经形成12a和12b的进一步证据是在脱卤化氢反应混合物的13C NMR光谱中出现了它们的所有13C信号(由于重叠的脂肪族区域中的一些信号除外)。
用强碱消除溴化氢通常通过具有离去取代基的反取向的E2机理进行(方案2)。比较起始溴化物6a和6b与产物7a和7b的比例(表1)以及7a和7b的异构体比例与溶剂极性变化的独立性,为我们的E2机制提供了支持。案件。因此,预期(9R *,10R*) - 6a可得到9-顺式-13-顺-7a,而9-反式13-顺式-7b应由(9R *,10S *)- 6b形成(方案2))。此外,溴化物6和11的比例与作为脱氢溴化反应异构体的13-顺式-7至13-反式12P(CH3NCH2CH2)3N的比例合理相关,支持11是主要的结论,如果不是鞋底,后者化合物的来源。
方案2
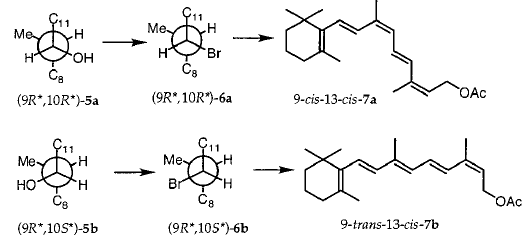
方案3
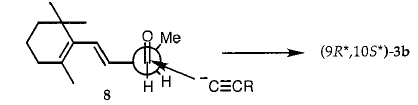
将5a和5b中的OH转化为6a和6b中的溴化物(方案2)无疑涉及离子中间体25,因为观察到形成大量重排的溴化物11并且因为在5a和5b中发生C10的部分差向异构化。与5a和6a以及5b和6b的1H NMR图谱非常相似,表明6a是纯5a的溴化的主要产物,而6b主要由纯5b形成。因此,在我们的情况下,三溴化磷将仲二烯丙基醇5转化为溴化物6,具有显着的构型保留。为了支持这一结论,建立了非对映异构醇3a和3b和/或4a和4b中的相对构型。首先,我们考虑将(Z)HC≡C(CH3)=CHCH2OH加入到2的羰基中的不对称诱导。当从Felkin-Anh模型预测的构象中受阻较小的面发生亲核攻击时,26其中将2-(2,6,6-三甲基环己烯-1-基)乙烯基视为L取代基,然后(9R *,10S *) - 3b占优势(方案3)。虽然这种添加中的非对映异构体过量(de)仅为中等(20%),但在结构紧密相关的2-取代丙醛与不同乙炔的锂或镁盐的反应中,27-32 de的值从未超过33%,并且配置发现主要产品与Felkin-Anh模型的预测完全一致。
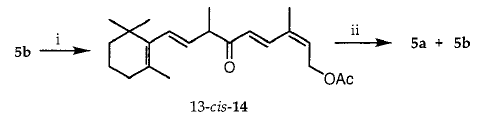
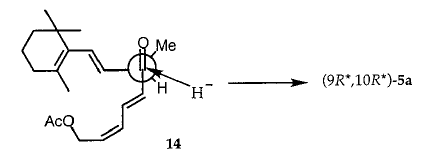
3b中的(9R *,10S *)构型以及4b和5b的另一个论点是从通过乙酸酯5b的PCC氧化33制备的酮14的还原的立体化学获得(方案4)。因此,当在0℃下用NaBH 4还原不稳定物14时,形成70:30(9R *,10R *) - 5a和(9R *,10S *) - 5b的混合物。文献中的先例报告通过引用Felkin-Anh模型结合ð* CdO-ð* CdC有吸引力的相互作用来稳定过渡态,从而使结构相关的R-甲基-α-ε-不饱和酮的氢化物还原的立体化学合理化。亲核加成。应用该模型(方案5)来减少14使(9R *,10R *) - 5a的形成合理化为主要产物。据报道,酮13-反-14可以用NaBH4和AlH(OtBu)3还原,但没有公开反应的非对映选择性。
方案4
方案5
非对映异构体8a和8b中的11,12-顺式构型是从H11-H12偶联常数(11.2Hz)的较低值推导出的,与相应的11,12-反式异构体4a和4b(15.6Hz)中的相比较。混合物中异构烃9的结构由1H和13C NMR光谱数据推导,包括1H-1H COSY实验。因此,13C NMR谱清楚地显示了111.8-114.5ppm区域中的末端亚甲基碳的四个信号,并且它缺乏与氧连接的碳的信号。二维1H-1H相关性允许我们将1H共振分配给混合物9a和9b的两个主要成分,分别具有10,11反式构型(JH10-H11)15.0和15.4Hz。 9a中C12-C13键周围的取代基的顺式排列可以基于H14相对于9b中的0.5ppm低场位移来指定。该结论预期分别来自化合物对9a和9b以及7a和7b中的C9-C15和C13-C7亚基的结构相似性。 MS数据支持9的配方。 CI-MS谱显示分子离子的突出峰和另外两个由两个烯丙基位置的断裂引起的峰。
尚未研究形成异构烃9的机理,但是当用氢化铝锂长时间处理时,8和4都不能提供可检测量的9。另一方面,当用LiAlH 4还原3的15个四氢吡喃衍生物时,在粗产物中发现9种全部四种异构体。
总之,发现(9R *,10S *) - 13-顺式-11,12-二脱氢-9,10-二氢-10-羟基亮氨酸(3b)是添加(Z) - 的主要产物。 LiC≡C(CH3)d=CHCH2OLi为(E)-2-甲基-4-(2’,6’,6-三甲基-1-环己烯-1-基)-3-丁烯醛(2)的羰基。在PBr3存在下,二醇(9R *,10R *) - 4a和(9R *,10S *) - 4b的15-乙酸酯转化为13-顺式-10-溴-9,10-二氢精氨酸乙酯(6)具有显着的配置保留。乙腈中的Proazaphosphatrane 1比DBU和DBN更快地从维生素A前体6和11中消除HBr。
实验部分
除非另有说明,否则材料是从商业供应商处获得的并且未经纯化而使用。溶剂是试剂级,在分子筛上预干燥,并且必要时在使用前从钠 - 二苯甲酮羰基中蒸馏(THF,乙醚)。通过施加真空并在室温下磁力搅拌直至达到20毫托,从油性反应产物中除去溶剂。但是,仍然存在残留的己烷,特别是对于粘性化合物,从它们的1H NMR光谱判断。通过在真空下加热除去己烷的努力导致分解。
1H NMR光谱在Nicolet NT-300和Varian VXR-300NMR光谱仪上在氘代氯仿中测量,而13C和31P NMR光谱分别在Varian VXR-300NMR光谱仪上在氘代氯仿和氘代乙腈中记录。使用氯仿(1H,7.23ppm)和氘代氯仿(13C,77.07ppm)共振作为二级标准,以四甲基硅烷的低磁场百万分率报告化学位移。 1H和13C化学位移分配由作为混合物的2,4b,8a,b,6和11以及7a和7b的混合物进行的2D 1H-1H相关性以及记录的2D 1H-13C相关性得到支持。图4b,8b和7a和7b的混合物。使用标准COSY和HETCOR实验在Varian VXR 300光谱仪上获得二维光谱。在Finnegan 4023质谱仪上测定9的CI-MS谱。
对于制备色谱分离,使用硅胶(60-200目,EM Science),除了维生素A异构体,其中仅发现失活的氧化铝(中性,Baker)有用。使用预涂有硅胶(IB-2和IB-F)或氧化铝(IB-F)的板(来自Baker的Bakerflex)进行TLC分析。使用以下溶剂体系:3:1己烷 - 乙酸乙酯(v / v,对于硅胶板)和10:1己烷 - 乙酸乙酯(v / v,对于氧化铝板)。
制备三甲基锍甲基硫酸盐(88%收率;点燃,40 70-75%收率),2-甲基-2 [2’ - (2’’,6’’,6’’- 三甲基-1,6-环己烯) -1’’基 - 乙烯基]环氧乙烷(产率98.3%,产率8.94%),和(RS) - (E)-2-甲基-4,2’,6’,6’ - 三甲基-1’根据所引用文献中的方法制备yl-基)-3-丁烯醛(2)(定量;产率85%产率)。
13-顺式-11,12-二脱氢-9,10-二氢-10-羟基苯乙醇(3)。顺式-2-甲基-2-戊烯-4-基-1-醇的二锂盐由醇(3.715g,38.6mmol)和正丁基锂(30.9mL,2.5M己烷溶液)在四氢呋喃(100)中制备。 mL)低于-15°C。在-15℃以下搅拌1小时后,加入室温的2(5.63g,27.3mmol)的THF(20mL)溶液,同时将反应烧瓶温度保持在-15℃以下。将反应混合物在该温度下搅拌1小时,然后将其温热至室温。加入冷水(100mL)后,用乙醚(2×200mL)萃取产物。将有机相用盐水洗涤至中性,并用MgSO 4干燥。蒸发溶剂,真空蒸馏出未反应的醇(0.2托,浴95℃)。留在烧瓶中的黄色油(7.046g,85%)通过NMR光谱法鉴定为3a和3b的足够纯度(TLC)的混合物,用于下一步骤。样品在硅胶柱上用己烷 - 乙酸乙酯(10:1至3:1,v / v)纯化,得到纯的3a和3b(2:3混合物),为无色油状物。
用LiAlH4还原3。向在氩气下冷却至2℃的LiAlH 4(1.736g,45.75mmol)在乙醚(75mL)中的悬浮液中,以1:1的速率滴加3(7.05g,23.3mmol)的乙醚(25mL)溶液。足够慢以使反应混合物的温度保持在6℃以下。然后将混合物在室温下搅拌16小时。冷却至-10℃后,缓慢加入水直至剧烈反应停止。将反应混合物用稀H 2 SO 4在0℃酸化,分离醚层,水相用乙醚(2→50mL)萃取。将合并的有机相用水,NaHCO 3水溶液洗涤,然后再用水洗涤至中性,用MgSO 4干燥并蒸发。将残余物在室温下真空(0.1托)保持48小时,得到6.779g非常粘稠的黄色油状物,将其在硅胶(50g)上用己烷和乙酸乙酯混合物进行色谱分离,得到异构体9的混合物(油状物,0.315g,5%),非对映异构体8的混合物(0.898mg,12.6%),部分分离的非对映异构体4a和4b的混合物(2.224g,31.3%)和结晶4b(1.503g,21.2%)。溶剂蒸发。将非对映异构体8在硅胶上进一步色谱分离,用10:1己烷 - 乙酸乙酯(v / v)洗脱,得到8a的纯样品和极性更强的8b。
单乙酸酯5a和5b的合成。将4b(1.397g,4.588mmol),2,4,6-可力丁(3.03mL)和乙酸酐(0.87mL,9.17mmol)的混合物在室温下放置48小时。用乙醚(50mL)稀释后,将混合物再次用冷的稀硫酸,水,NaHCO 3水溶液和水洗涤,然后用MgSO 4干燥。用10:1和5:1己烷 - 乙酸乙酯(v / v)在硅胶上进行色谱分离,得到二乙酸酯10b(174mg,9.5%,油)和5b(1.440g,89.5%,油)。以类似的方式,将4a和4b的混合物酯化。用己烷/乙酸乙酯混合物(100:5至2:1,v / v)在硅胶上进行色谱分离,得到5a和5b的部分分离。
三溴化磷与5的反应(通用程序)。在-20℃和氩气下,向5(10mmol)的乙醚(10mL)溶液中注入PBr 3(12mmol)。将反应混合物搅拌1小时,同时使其在此期间达到室温。然后将其用乙醚(40mL)稀释,用冷盐水,NaHCO 3水溶液洗涤,然后用盐水洗涤至中性并用MgSO 4干燥。蒸发乙醚后,将粗产物真空(0.02托),得到6a,6b和11的混合物,为黄色油状物,产率85-95%。当在室温下放置时,该材料缓慢变为棕色,因此在下一步骤中立即使用。
用1,DBN或DBU(一般程序)从6a,6b和11的混合物中消除HBr。将先前实验中获得的6a,6b和11的混合物溶解在苯或甲苯(1mmol,1mL)中,并与1.2当量的1,DBN或DBU一起回流。或者,将溴化物的混合物溶解在乙腈(1mmol,在1mL中)中,并在室温下与1.2当量的1,DBN或DBU一起搅拌。将反应混合物用乙醚稀释,用盐水洗涤至中性。将粗产物通过失活的氧化铝(12g,5mmol)过滤,得到7a和7b(主要)和12a和12b(次要)的混合物。
合成酮14.将乙酸酯5b(0.193g,0.55mmol)溶解在CH 2 Cl 2(2mL)中,然后在0℃下分批加入PCC(0.178g,0.83mmol)。在0℃下3小时后,加入己烷(10mL),用移液管取出有机溶液并浓缩。将黄色残余物在硅胶(5g)上用己烷 - 乙酸乙酯(20:1,v / v)色谱分离,得到4(60mg,31%),为淡黄色油状物。在NMR光谱的基础上(参见支持信息),材料被判定为ca. 纯度90%。
将酮14还原为14(77mg,0.22mmol)在CH 2 Cl 2和甲醇的1:1混合物中的溶液,在0℃下分批加入NaBH 4(8.4mg,0.22mmol)。 30分钟后,加入己烷(20mL),然后加入H 2 SO 4水溶液(6%)(0.1mL)。然后除去冰水浴,用MgSO 4干燥反应混合物。将粗产物在硅胶(5g)上用己烷 - 乙酸乙酯(10:1,v / v)色谱分离,分别得到5a和5b的7:3混合物。比较5a和5b中的H3C16,17,H3C19,H3C18,H3C20,HC10和HC12的1H NMR积分并取平均值。
原文标题:《Micro-batch reactorfor catching intermediates and monitoring kinetics of rapid and exothermichomogeneous reactions》
原文出处:ChemicalEngineering Journal 214 (2013) 149–156
涉及维生素A衍生物


目前评论: