- A+
摘要:镁金属与有机卤化物RX在醚溶剂中的反应可能是典型的金属腐蚀,在这种腐蚀中,Mg2+的稳定基本上是通过其与溶剂的配位,驱动其从金属中损失,从而减少Rx和反应中间物,如金属表面的R•的减少。尽管卤代烷通过中间自由基在溶液中扩散的非链机制形成格氏试剂,但在某些乙烯基和芳基卤化物的格氏反应中,即使中间自由基R•会很快发生异构化,也会发生非常少量的自由基异构化。这表明这些乙烯基和芳基卤化物的主要非自由基机制或中间自由基R•的寿命极短的机制。由于前者似乎更可能,因此提出了一种通过过渡状态[RX2−]的离子机制。多晶镁的表面研究表明,“氧化物”层主要是Mg(OH)2,并且是机械钝化。在没有促进剂的情况下,格氏反应发生得非常缓慢,直到有足够的Rx渗入镁表面,并在那里发生反应,使Mg(OH)2层剥落。
关键词:格氏试剂;镁;腐蚀;还原;烷基卤化物;环丙基卤化物;乙烯卤化物;芳基卤化物;氧化镁;氢氧化镁
1.引言
在“格氏反应”中,格氏试剂RMgX在适当的溶剂SH中形成,通常是二乙醚(DEE)或四氢呋喃(THF),来自镁金属Mg和有机卤化物RX。副产物可包括RR,RH,R(-H),RS,SS,S(-H)和MgX2。在过去十年中,Hamdouchi和Walborsky(HW)以及Garst和Ungváry(GU)对Grignard反应机制进行回顾。Kharasch和Reinmuth(KR)对该主题的早期历史进行了评述。
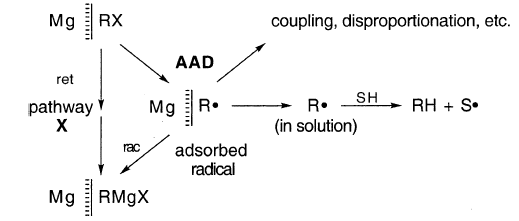
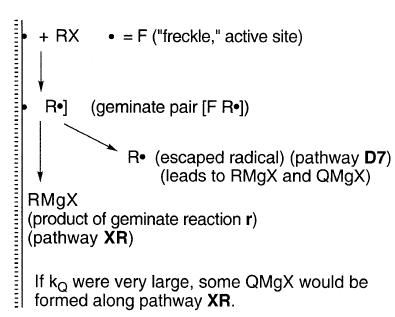
HW和Gu同意一个普遍接受的观点,即“格氏基团”R•是沿着有时是单独或主要的R途径的中间产物。

沿R途径,中间基团R•被受还原(r),偶联/歧化(c),对醚溶剂的攻击,或其他一级或假一级反应(q),如异构化或捕获,导致RMgX和副产品的形成。链式反应被自由基捕获和其他结果所排除(Gu,235)。
在细节上,HW和Gu有显著差异。HW接受了几乎所有R•都保持吸附在镁表面MgZ上的假设。GU不认同这些假设。
HW坚决主张像AAD这样的机制。这是“AAD”,因为沿着通道rcs,按照这个顺序,r•在MgZ处R和C保持吸附(“a”),但在s处扩散未稀释(“d”)。
其他可能性包括ADD和DDD。DDD受到GU的青睐.
根据关于在反应表面附近扩散的自由基行为的假设,HW认为某些数据与DDD不一致。Gu回应说,这些假设是无效的,HW引用的数据实际上与DDD一致。环丙基溴的反应数据不仅与DDD一致,而且与AAD和ADD(Gu,216–218)也不一致。
如果这是整个故事,这件事可能会被认为是解决了。然而,异构化研究的结果使情况复杂化。
一些中间基团R•可以异构化。
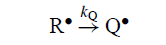
在无限稀释时,自由基偶联不重要,异构化的程度由异构化速率常数kQ和R•的寿命决定,受限于与异构化反应r和s竞争的反应。有时在RMgX中异构化程度如此之低,DDD只有在R•具有极短的r限制寿命τR,10-10s或更低(GU,218-219)时才能解释它。相比之下,对这些或类似情况的产品分布的其他方面的动力学分析表明寿命更长,τR≈10-7s。
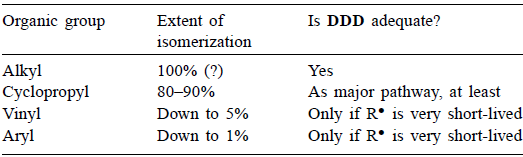
大体上,情况按有机组R的性质分类,见表1。
表1
提出了两个主要问题。
(1)中间基团R•是否仍然吸附在MgZ上?对于烷基和环丙基的情况,现在很清楚R•在MgZ附近的溶液中扩散。虽然乙烯基和芳基的情况没有具体证据,但没有理由相信它们与烷基和环丙基的方式不同。
(2)为什么部分保留配置?这个问题尚未得到明确答复。必须有一条路径X,沿着这条路径(a)一些中间基团R•具有如此极短的寿命,以至于它们保持其构型或(b)R•不是中间的,R保留其构型。
途径X尚未定义。
这些问题涉及有机机制,它将R从RX跟踪到RMgX。无机机制将来自金属的Mg描绘成RMgX。尽管研究很少,但与其他金属腐蚀反应类比,可能会引发合理的推测。
诱导期是该机制的另一个方面。通常,Grignard反应不会立即在混合试剂上开始。相反,启动需要一段时间。为什么?诱导期间会发生什么?
至少部分答案可能涉及“接收Mg”时涂覆MgZ的“氧化物”层,“从瓶子外面”,或者在暴露于大气之后。“氧化物”层主要由Mg(OH)2(GU,255)组成。该层使镁具有暗淡的外观,但在正在进行的格氏反应中,MgZ通常会获得金属光泽。
本次评述有三个考虑因素。首先,HW和Gu试图做到全面和详细。我们更具选择性,更多示例、细节和参考资料请参见HW和Gu。第二,考虑新的发展。第三,本文首次对“氧化物”层的性质及其与各种试剂的相互作用的最新结果进行了评述,其中一些试剂通常用于在格氏反应中“激活”镁。
JFG是第2节“格氏反应”的作者,MPS是第3节“氧化层和诱导期”的主要作者。
2.格氏反应
2.1. 溶剂
格氏试剂可以在各种非质子溶剂中制备,包括叔胺。即便如此,两种醚中的一种,即DEE和THF,几乎总是在实践中使用。它们分别具有介电常数4.3和7.4。
在二恶烷中,介电常数为2.2,反应通常不能引发。此外,MgX2不溶。将RMgX溶液倒入二恶烷中将沉淀MgX2,使R2Mg保持在溶液中。
1,2-二甲氧基乙烷(DME,甘醇二甲醚),介电常数7.2,是有用的,但有时存在溶解度问题。此外,当希望水性和有机层干净地分离时,DME的高水溶性会妨碍后处理程序。
对于大规模工业过程,挥发性和易燃的醚DEE和THF存在安全隐患。“丁基二甘醇二甲醚”,CH3CH2CH2CH2OCH2CH2OCH2CH2-OCH2CH2CH2CH3,是格氏试剂制备和反应的优异溶剂。其闪点为118℃(DEE:-45℃;THF:-14℃),其水溶性非常低。它的高沸点256℃(DEE:34℃;THF:66℃)和闪点允许比DEE或THF更高的反应温度,这些可以导致产生耐受的格氏反应后者的溶剂。
2.2. 配位作用
对于格氏试剂,其他有机镁化合物和卤化镁,已经确定了许多晶体结构。许多格氏试剂RMgX和二有机镁化合物镁化合物R2Mg在扭曲四面体的中心以四配位Mg结晶:
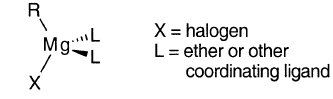
如果配体可以匹配,Mg可以是五配位的:
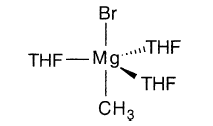
溴化镁已从THF中以4配位和6配位形式结晶:
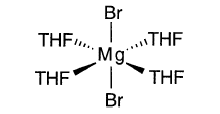
这些结构强调了配位溶剂在稳定格氏试剂和卤化镁中的作用。
与Mg2 +的溶剂配位还必须提供大量的热力学驱动力以减少Mg,包括RX在醚溶剂中形成格氏试剂的那些。在格氏试剂形成过程中,Mg从金属状态(基本上未溶剂化)进入基本离子状态Mg2 +,在此状态下,Mg与醚溶剂分子以及配体R和X强烈配位:
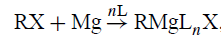
L=配位溶剂分子
在任何特定情况下,结构的许多细节决定了Schlenk均衡的位置:
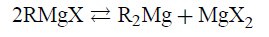
类似的细节决定了其他平衡的位置,涉及聚集,复合离子形成和各种物种的溶解度。这些细节包括在与Mg 2+配位层中起作用的空间效应。虽然现代计算工具原则上提供了理解这些和其他对格里纳德均衡的影响的手段,但是没有实现这种理解。
2.3. 腐蚀,•MgX和RMg•
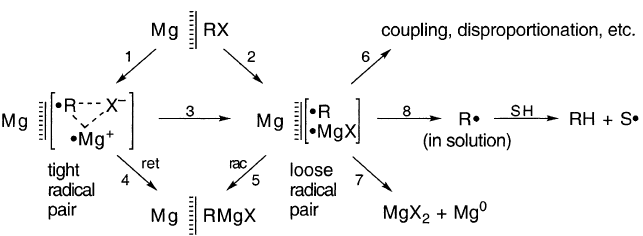
Grignard反应的一个基本机制的简单表述为引发了一个中间产物•MgX:
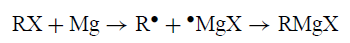
尽管该方案在文献中很受欢迎,但没有证据支持它。实际上,证据反对它。 CIDNP净效应来自具有不同g值的自由基的反应。如果RMgX由成对[R••MgX]形成,则可以预期它们,但事实上只发现由对[R•R•]产生的多重效应。没有证据表明中间体RMg是R•与Mg反应的可能产物:
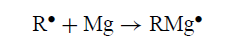
典型的•MgX或RMg•作为中间体的调用并不能解释Mg如何离开金属晶格。这个想法似乎是由RX或R•简单地拔出来的。虽然这是可能的,但它不符合通常的金属腐蚀机理。
如果没有相反的证据,推测Grignard反应遵循与其他金属腐蚀类似的机制似乎是明智的:
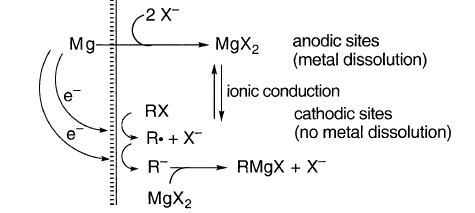
这里,RX和R•的减少是由于Mg在阳极位置溶解Mg2+所致。如果没有发生减少,则Mg将留下过量的电子,也就是说,它最好表示为Mg-f,其中f是一些小的值。这种金属离子的溶解是与水接触的金属熟悉的半电池电位的原因。
在水中,离子传导是容易的,并且即使在宏观尺度上,腐蚀过程的阳极和阴极位置也可以很好地分离。当液体的极性小得多时,例如醚,离子传导很慢,阳极和阴极位置必须靠近在一起,甚至可能在分子尺度上。否则,由于不同的电荷分离的发展,腐蚀反应很快就会停止。
通常,金属的腐蚀遵循由其晶格结构确定的几何图案。失去溶液的原子往往是那些与晶格最少结合的表面原子,例如角原子。由于这种情况反复发生,腐蚀沿着晶面进行。通过光和原子力显微镜(GU,253-255)观察到Grignard反应中的Mg。通过这种方式,格氏反应符合其他金属腐蚀。
对模式腐蚀的观察排除了非选择性拔掉。然而,选择性拔掉仍然可行。即使如此,也没有理由怀疑格氏反应不是典型的(非透明的)金属腐蚀。
2.4. 极性溶质
人们早就知道MgX2催化格氏反应(KR,8-11)。因此,在DEE中MgBr 2的存在可以消除在环丙基溴,CpBr的格利雅反应中观察到的诱导期。 [注意:此处和整个过程中,“Cp”代表环丙基,而不是环戊二烯基。]当最初不存在MgBr2时,该反应呈现“S”形进展曲线,这是自催化的特征(图1)。
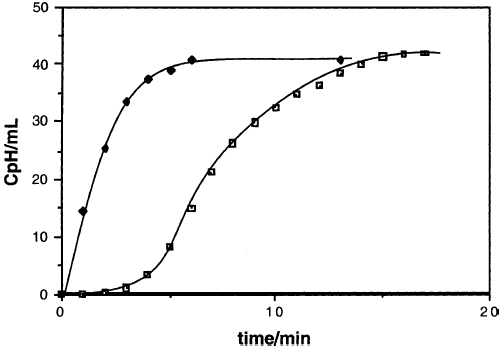
图1
由于MgBr2伴随其他副产物,它可能是自催化产物。然而,由于其他极性添加剂也可以促进反应(GU,257-259),RMgBr也可以起作用。
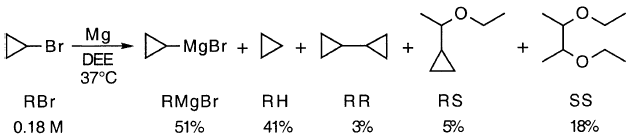
诸如I2和BrCH2CH2Br的试剂通常用于促进格氏反应。已经提出这些试剂通过在与MgZ反应中蚀刻来激活MgZ。蚀刻假设的具体测试表明它不是一个因素。在溶剂中包括Mgl 2或MgBr2最初具有与包括I2或BrCH2CH2Br相同或更好的效果,无论所用的MgZ是否先前已经“蚀刻”。
通过“夹带”引发了不情愿的反应,即通过在反应混合物中加入反应性烷基卤以及寻求格氏试剂的不含卤化物(KR,38-45)[17,18]。现在看来,夹带仅仅是引入RMgX和MgX2的方法,极性溶质促进了不情愿的卤化物(GU,259)的反应。
尚未发现极性溶质促进格氏反应的具体作用方式。
2.5. 烷基卤化物
2.5.1. 烷基自由基中间体的证据
烷基自由基中间体的证据是压倒性的。它包括以下事实:(a)副产物RR和RH+ R(-H)以特征自由基偶合/歧化比形成;(b)发生特征性的自由基异构化; (c)当存在极端陷阱时,以RMgX为代价形成诱捕产物;(d)RMgX及副产品展示CIDNP。溶剂侵蚀的观察结果也是自由基中间体的证据,但在烷基情况下几乎没有溶剂侵蚀。
KR回顾了关于中间自由基R•的假设的早期历史。在20世纪20年代推测,格氏反应是由Mg和MgX2形成的MgX引发的自由基反应。到1954年,通道R被普遍接受,主要是因为发现RMgX与RX的反应太慢,在大多数情况下,是因为副产物RR和RH + R(-H),导致自由基偶联/歧化作为最合理的替代方案。
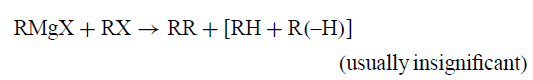
RR与RH或R(-H)的比率与初级,二级和三级自由基反应的预期值相匹配,与表征SN2 / E2反应的那些非常不同。这很重要,因为否则RR和RH + R(-H)可能来自与中间碳负离子R-或碳阴离子物种的RX反应,有证据表明:
原则上,可以研究光学活性烷基卤化物的格氏反应。外消旋产物可以反映R•的外消旋化。在这些研究中发现了外消旋产物(HW,155-159),但这可能是由于RMgX的外消旋化,但尚未排除。
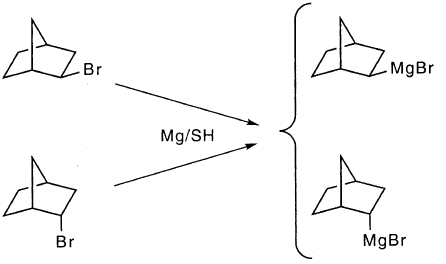
尽管外- 和内 - 降冰片基溴化镁充分保持其构型,但已经研究了外 - 和内- 前 - 溴代溴化物反应的两组获得了相反的结果。从异构反应物获得相同的,不同的产物分布。这需要澄清。
中间伯烷基被DCPH捕获,效率低(~25%),TMPO•有效(高达95%)。TMPO捕获的高效率表明R是唯一途径。
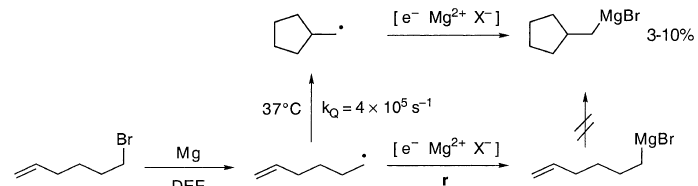
CIDNP在RMgX中找到。在镁与乙基碘在二正丁基醚中的反应中,在NMR管中进行,并且在CH3CH2MgI的CH2的NMR信号中观察到E/ A多重效应。这是CH3CH2MgI的预期相位,如果由自由基CH3CH2•形成,它们从随机自旋对[CH3CH2••CH2CH3]中逃逸出来,这是在独立扩散自由基彼此相遇时产生的。环化自由基探针如5-己烯基表示自由基中间体。5-己烯基卤化物的格氏反应得到(环戊基甲基)卤化镁,收率为3-10%。(5-己烯基)卤化镁环化太慢而不能解释这些观察结果。
一种降冰片烯基乙基自由基探针,其环化速率常数kQ值为〜107s-1,得到的环化和非环化格氏试剂的产率几乎相等:
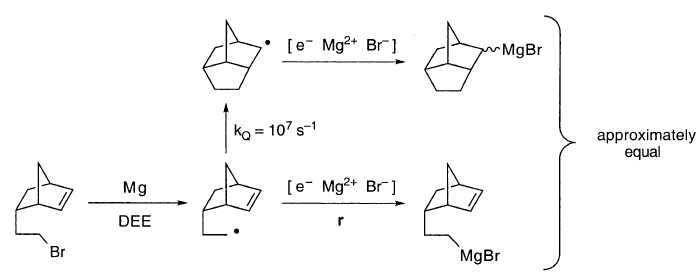
于环化和r直接竞争,这表明R•的r-限制寿命ΔR是~10-7s。
2.5.2. AAD
在Grignard反应中,c的副产物通常比s的副产物占主导地位。认为甲基自由基不可能离开MgZ,在溶液中扩散,并且在没有发现的情况下经历c,所以KR(63)表明中间基团R•可能保持吸附在MgZ上,具有足够的流动性,在那里彼此相遇并经历C。这是AAD。
然而,KR(63)也评论说,“自由基是否真正进入大量解决方案体内是一个几乎无法得到任何保证的问题”。他们继续建议(KR,64)“反应性较低的自由基(即不能从溶剂中提取氢原子的那些)可能会在系统中累积,直到它们彼此反应或与格氏试剂呈现出相当大的比例。 ”。显然,该组包括除甲基以外的烷基。相应的机制是ADD和DDD。
接受甲基的KR位置的人可能会问:“为什么其他烷基不同?”找不到令人满意的理由,那个人可能喜欢AAD用于所有烷基卤化物的格氏反应,假设在体系中积累了格里纳德自由基, “正如KR所描述的,实际上是在MgZ。
2.5.3. DDD
Lawrence和Whitesides指出,衍生自典型烷基卤化物的格氏反应基团与溶液中的相同基团非常相似。除了可捕获之外,如“溶液”自由基,它们经历特征性的自由基异构化,产生CIDNP,并且在特征比率上偶然和不成比例。吸附不太可能不影响这些比例。这些事实强烈建议扩散Grignard自由基。
分子间诱捕表明DDD。通常认为,捕获主要发生在溶液中,而不是MgZ。因此,观察到DCPH的低效捕获和TMPO的高效捕获•指向扩散中间体R•。
尽管HW(203-205)接受了DCPH结果,但他们批评TMPO•实验的理由是,除了R•与TMPO•之外的反应中可能出现明显的捕获产物。然而,TMPO研究的作者花了很大力气才找到抑制其他反应的条件。此外,由于预期DCPH的捕获效率较低,因为它的反应性低于TMPO•,因此没有明显的理由怀疑这两组捕获结果不相互一致。
在没有显着性的情况下,对于烷基卤化物的格氏反应,难以设想AAD的测试,其支持很少的确定预测,因为吸附的自由基的行为未被指定和未知。补充的临时假设可以使其与许多没有主动预测的结果(HW,GU)一致。然而,这些临时假设中的一些是相互矛盾的。
相比之下,DDD具有相当大的预测能力。它可以通过产品分布的动力学分析进行测试。其对烷基卤化物反应预测的成功使DDD成为首选理论。
在下文中,MgZ被视为均匀反应性的无限平面。因此,R•的唯一显着扩散是在x方向上,垂直于MgZ。
对于可以进行Q•的一级异构化的Grignard自由基R•,DDD如何预测Q•形成的程度将随kQ变化,其他因素保持不变?
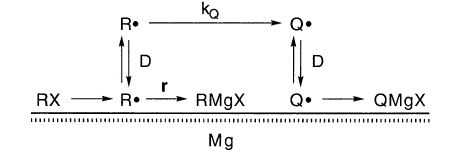
一个单纯的答案可能会假设r可以表示为一阶反应,rrate =τR-1 [R•]。然后QMgX / RMgX将与kQ成正比,这是不正确的:
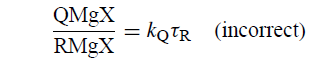
缺陷在于扩散R•仅经历R(时变)的t(时变生存概率s(t)不是指数衰减:
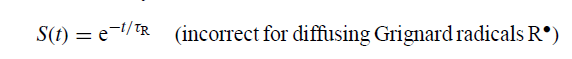
相反,溶液中的扩散与表面反应的混合导致复杂的衰变定律,
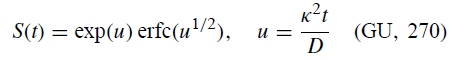
其中κ是r的非均匀速率常数,[R•]0是Mg•(x = 0)处R•的溶液浓度:
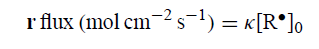
因此,即使忽略c,获得将q[QMgX / RMgX]与kQ相关的表达也是微不足道的。(无限稀释时不会出现c,因此这是“无限稀释”情况。)正确的结果是“平方根法”(GU,202)
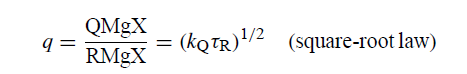
如果τR在一系列情况下是恒定的,那么屈服比将与kQ的平方根成比例。也许,扩散扩散遵循t1 / 2定律并且kQ的单位是反时间是相关的。
对数 - 对数图将具有斜率1/2:
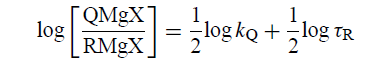
首先测试了这个平方根法则,如图2,三个异构化的烷基格氏离子基团,具有已知的kQ值(5-己烯基:1.8×105s-1; 正二甲基乙基:1×107 s-1;环丙基甲基:1×108 s-1)。
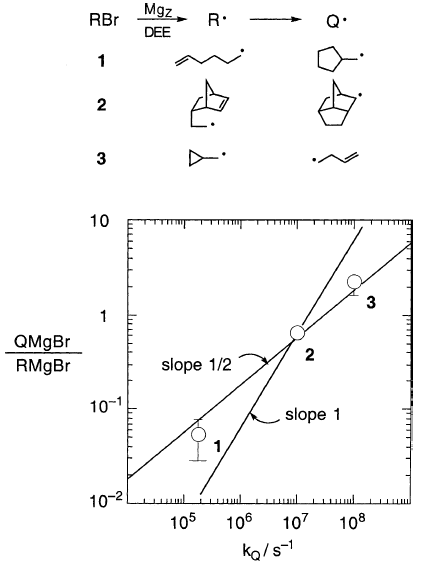
图2
拟合是令人满意的,比斜率1的线要好得多,斜率线1由一阶竞争的初始假设预测,QMgX / RMgX =kQτR。当考虑c的出现时,获得更好的适合1。
Whitesides收集并建立了许多其他例子,其中Grignard自由基的一阶或伪一阶过程与它们的还原为RMgX竞争。图3显示,观察到的RMgX的分数产率A与通过平方根法预测的那些合理地一致。
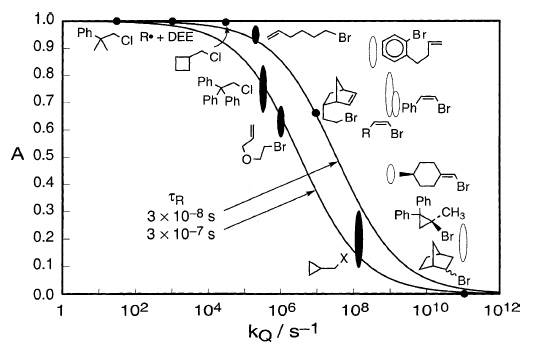
图3
虽然烷基壳(固体椭圆形)很合适,但环丙基,乙烯基和芳基(开口椭圆形)不适合,这表明后者具有不同的κ值或遵循与前者不同的机制。
使用DCPH和TMPO•的捕获实验的结果可以类似地绘制,将捕获作为伪一级处理(图4)。
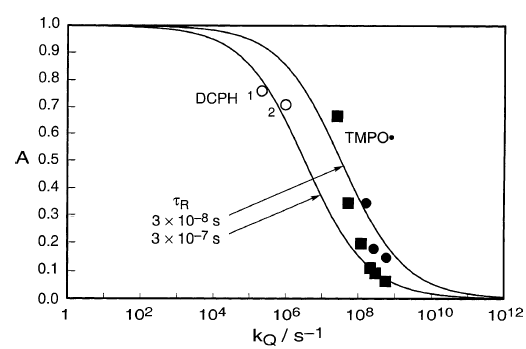
图4
两组捕获数据DCPH和TMPO•与图3的平方根法相关性一致,表明它们是同等有效的陷阱,尽管上面提到的由HW表示的保留。
在理想情况下,DDD得到了扩散反应方程的精确解,其中类似反应的速率参数(κ,kS,kC)具有相同的值:
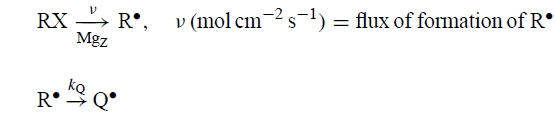

当溶剂侵蚀和异构化都很显着时,完整的产物分布由三个复合无量纲参数V,Δ和G2 - 1的值决定:
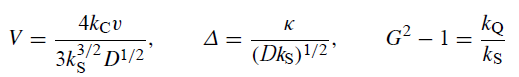
当异构化发生但溶剂侵蚀可忽略不计时,产物分布由两个参数VQ和ΔQ确定:
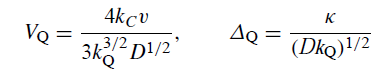
当发生溶剂侵蚀但异构化可忽略不计时,产物分布由V和Δ确定。当既不发生异构化也不发生溶剂侵蚀时,产物仅由RMgX和RR(包括歧化产物)组成,它们的分布由单个参数F确定:
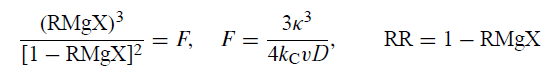
图5
图5(左)显示了计算的产物分布如何与在DEE中5-己烯基溴的格氏反应所观察到的相比。这些反应得到RMgBr [(5-己烯基)溴化镁],QMgBr[(环戊基甲基)溴化镁],RR(1,11-十二碳二烯),RQ[(6-庚烯基)环戊烷)和QQ(1,2-二环戊烷乙烷))。 RR,RQ和QQ原则上包括歧化产物,但这些没有报道,并且对于伯烷基是次要的。未检测到溶剂侵蚀产物。
在不调整六个速率参数v,kQ,kS,κ,2kC和D中的任何一个的情况下获得DEE的计算值。每个都被指定测量值或典型值:v = 2×10 -5 mol cm-2 s- 1; kQ = 4.4×105 s-1; kS =4.4×103 s-1(测量为〜103 s-1);κ= 30 cm s-1; 2kC = 3×109 M-1 s-1(典型值); D = 3×10 -5 cm 2 s-1(典型值)。这些对应于相关无量纲参数的以下值:V = 25,000; VQ = 25.0; Δ= 82.6;ΔQ= 8.26; G2 - 1 = 100.所有值都非常靠近代表完美拟合的线。
IG,ID和H的值尤其值得注意。IG和ID分别是格氏试剂PMgBr(RMgBr+ QMgBr)和PP(RR + RQ + QQ)中异构化烷基Q(环戊基甲基)的百分比。对于DEE和THF中的反应,IG约为3%,而ID超过20%。如果R•和Q•仍然吸附在MgZ(AAD)上,则需要在表面MgZ处由不同的R•和Q•池形成PMgBr和PP。没有解释为什么或如何发生这种情况。
DDD提供了一种自然的解释- PMgBr仅在MgZ形成,而PP在任何地方形成。对于DDD,图6给出了R•和Q•的计算稳态浓度曲线。随着与MgZ的距离增加,P•中P•的比例稳定增加。因此,PMgBr形成在平面中,x= 0(MgZ),其中Q•的比例最小(~3%),但PP形成在任何地方,并且结合了更多的Q•。
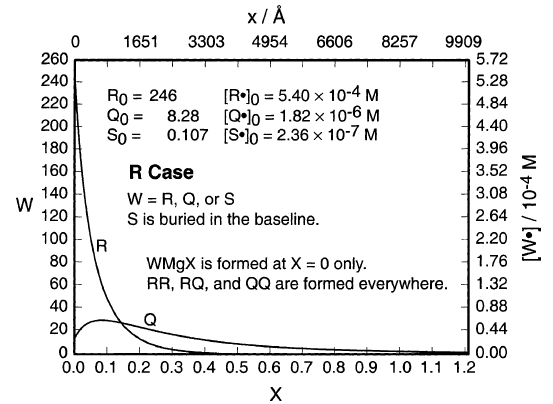
图6
H是收率RQ与RR和QQ的几何平均值之比。每当PP将R•和Q•与其种群成比例时,其值预计为2:
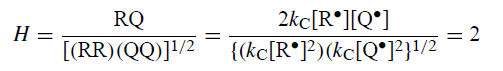
观察值接近1并且与计算值合理地一致。AAD预测H = 2,除非吸附的R•和Q•没有统计学上纳入PP。没有AAD解释为什么H = 2被提供。
DDD再次提供了一种自然而简单的解释。虽然在平行于MgZ的每个平面中形成的PP的H= 2,但观察到的PP在所有平面中形成,并且这些平面上的产量的总和改变了H的净值。
作为求和如何降低H值的一个例子,考虑在两个平面中形成的RR,RQ和QQ的可能分布:
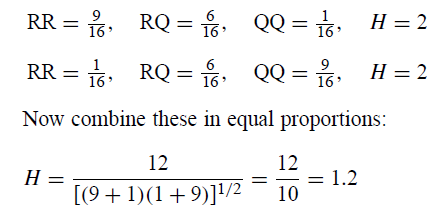
每个平面的H值为2,但组合值为1.2。
对于5-己烯基溴在THF,二正丁基醚(DBE)和二正戊基醚(DPE)中的格氏反应,调节速率参数以获得最佳拟合。图5(右)和7。
对于烷基卤化物的格氏反应,DDD预测所有已知的事实,包括IG和ID之间的差异以及H的异常观察值。对AAD或ADD没有有效支持。
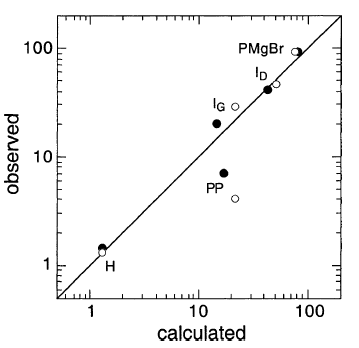
图7
2.6. 环丙基卤化物
2.6.1. 环丙基自由基中间体的证据
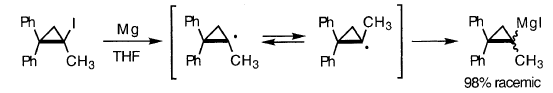
对于环丙基系统,中间体R的证据包括外消旋化,偶联/歧化和溶剂侵蚀。光学活性的1-甲基-2,2-二苯基环丙基碘化物产生98%外消旋的格氏试剂,即使格氏试剂在反应条件下是光学稳定的。外消旋中间体可能是R•:
其他1-甲基-2,2-二苯基环丙基卤化物提供更多的保留,但大范围的外消旋化仍指向沿主要途径的环丙基自由基中间体:
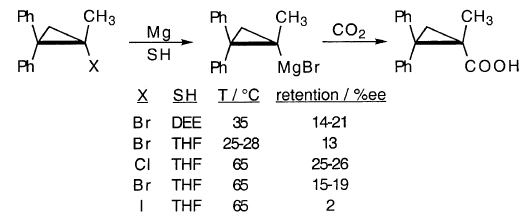
1-甲基-2,2-二苯基环丙基溴在DEE中的格氏反应得到c的产物,总产率为16%,4%RR和12%R(-H)(HW,160)。这相当于RH(R•R•)歧化反应收率为12%。由于RH的总收率为23%,RH的11%收率显然是由s或R•/S•歧化引起的.c和s的出现表明R•中间体。
环糊丙烯在DEE中的格氏反应给出了s的明确证据:
这些产品预计用于途径R.很难想象出合理的替代方案。
来自环丙基卤化物的Grignard反应而不是典型烷基卤化物的s产物的存在反映了环丙基在氢 - 原子转移反应中的更大反应性。对于典型的烷基,从醚溶剂中提取氢原子的拟一级速率常数kS为~103s-1,对于环丙基,其为~106s-1(GU,214)。
2.6.2.AAD
当它在溶液中由过氧化物产生时,1-甲基-2,2-二苯基环丙基R•攻击溶剂或环开到烯丙基Q•,然后偶合
到QQ。没有找到RR:
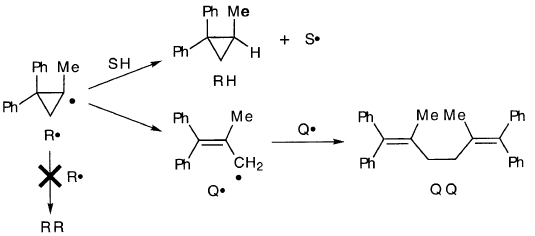
相反,1-甲基-2,2-二苯基环丙基溴在DEE中的格氏反应得到异构化的格氏试剂QMgBr,产率仅为1%。形成RR和RH + R(-H)(16%),但未检测到QQ,也未检测到RQ。
为了解释RR的形成以及Grignard反应中缺少QQ,Walborsky和Aronoff采用了KR的吸附假设(HW,160-164)。为了解释观察到的部分外消旋化和部分保留配置,他们提出了两种途径,即通过“松散基团对”[R••MgX]进行外消旋化,并通过“紧密自由基对”[RX•- •Mg +]进行保留。其中留在MgZ:
除了上图的外,未定义松散或紧密的自由基对。由于没有证据表明这些中间体,KRW机制是不必要的。它的基本特征保留在一个更简单的机制中,其中吸附的R•取代松散的自由基对,并且通路X通过紧密的自由基对取代反应。简化的KRW机制是由路径X增强的AAD:
HW引用了许多并行的例子。毫无疑问,Grignard和同类型的“溶液”自由基通常表现出不同的反应模式。这是AAD的原始论据。
另一种是基于激进的诱捕。对于环糊丙基溴在DEE中的格氏反应,Garst和同事发现(RMgBr)≈50%,通过滴定测定。根据Walborsky和Zimmerman的说法,同样的收益率为24%。在二环己基膦-P-d(DCPD)存在下,预期其是有效的自由基捕获剂,仅形成4%(RD)。根据这些作者的说法,诱捕的程度是微不足道的 - 很少有环丙基格氏离子被捕获,因为很少有人离开MgZ。
尽管这被认为是AAD最强有力的证据之一,但现在很明显报告的结果不完整,而且系统比Walborsky和Zimmerman假设的要复杂得多。下面讨论关于这些反应的其他结果。
2.6.3. DDD
对于s重要的格氏反应,DDD,ADD和AAD对降低kS值的影响做出了明确的预测。当溶剂氘化时,通过主要的动力学同位素效应,kS可能降低近6倍。由于在环丙基卤化物的格氏反应中存在显着的溶剂侵蚀,因此可以通过确定溶剂氘化对产物分布的影响来区分AAD,ADD和DDD。
在AAD中,吸附的R•的竞争性命运是r,c和解吸,其总是遵循s。氘代溶剂对决定产品分布的任何竞争过程都没有影响。因此,它对该分布没有影响。
在ADD中,r仅与R•的解吸竞争。在解决方案中,c和s竞争。氘代溶剂不会影响r,但会以s为代价增加c。产量RMgX将不受影响,但c产品的产量将以牺牲s的产量为代价而增加。
在DDD中,r,c和s彼此竞争。氘代溶剂会以s为代价增加r和c。产量RMgX和RR将以s的产品为代价而增加。
为AAD辩护的Walborsky和Aronoff的实验提供了该机制的第一个反证。由于其他原因,他们研究了溶剂过氘化对光学活性1-甲基-2,2-二苯基环丙基溴在DEE和THF中的格氏反应的影响,见表2。
即使观察到的微小变化都在DDD预测的方向上,但是对于氘化DEE的影响被认为是微不足道的,并且结果被认为是支持AAD。由于没有确定RR,它可能已经增加并且可能已经增加(参见下面的环丙基溴的相应结果)。如果是这样,结果可能与ADD一致。如果观察到的小的变化事实上是显着的,那么它们甚至可以与DDD一致。因此,DEE结果的重要性不是很明显。
表2
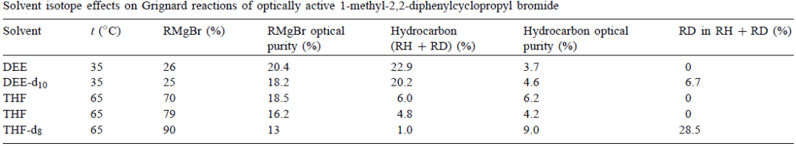
对于THF,结果很清楚。如DDD预测的那样,溶剂perdeuteration增加(RMgBr),代价是(RH+ RD)。这排除了AAD和ADD,两者都预测RMgBr将不受影响。由于DEE结果可能被解释为支持AAD,Walborsky和Aronoff选择忽略其THF结果的含义。他们没有评论他们解释的DDD性质,他们表示对kS的主要动力学同位素效应将自由基R•从s转移到r。
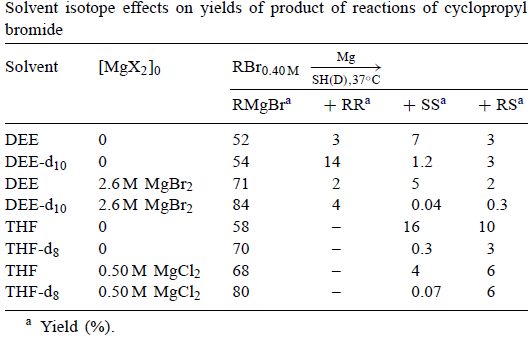
Garst等。确定了环丙基溴在未氘代和氘代DEE和THF中的格氏反应产物,结果总结在表3中。在所有四种情况中,s产物的产率受溶剂氘化的影响。这排除了AAD。对于没有添加MgBr2的DEE,RMgBr略有增加,RR的产量大幅增加。这与ADD最为一致,但如果RMgBr的增加是真的,也可能与DDD一致。
表3
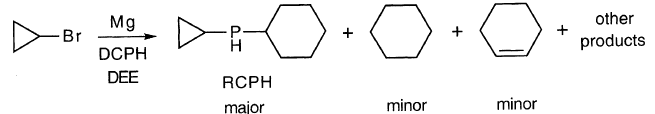
因此,似乎DCPH(D)捕获环丙基自由基的化学反应是不寻常的。它不是简单地从P-H中提取H•。
Garst和Ungváry发现DCPH是DEE中环丙基溴的Grignard反应中的有效自由基捕获剂,见表4。
表4
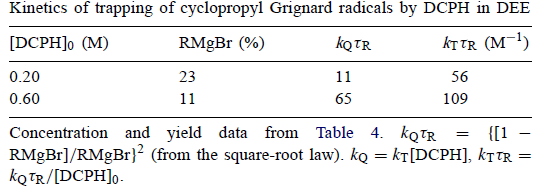
正如预期的那样,随着[DCPH]0的增加,s产品消失并且(RMgBr)稳定下降。此外,RCPH稳步增长。当发生这种情况时,RMgBr + RCPH的总和仅略有变化,为40-35-30%。显然,RCPH是R•捕获的主要产物。RH似乎是次要产品。与Walborsky和Zimmermann提供的解释相反,DCPH是环丙基格氏反应基团的有效陷阱。
根据平方根定律,kQτR= {[1 -RMgBr] /RMgBr} 2。将其应用于表4的条目给出表5中的结果。
表5
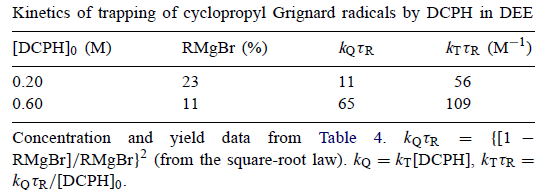
在最后一列中给出kTτR的计算值,其中kT是通过DCPH捕获环丙基的二级速率常数,kQ = kT [DCPH]。原则上,kTτR的值应该是恒定的。这些估计是通过将kQτR除以[DCPH] 0,即通过将[DCPH]近似为其初始值的常数而获得的。当然,这不是真的,但是当[DC] 0超过[RBr]0时,它是更好的近似值。即便如此,kTτR的计算值仅相差两倍。如果109是kTτR的更好估计并且如果τR=1×10-7s,则kT = 1×109M-1s-1。如果τR=3×10-8s则kT = 4×109M-1s-1。接近扩散控制极限的这些值似乎合理,比伯烷基和DCPH的kT大约三个数量级。
产物分布的动力学分析为甲基丙基溴的格氏反应提供了DDD的进一步测试,图8。
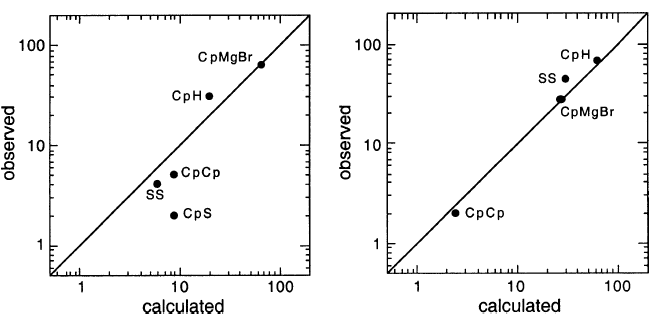
图8
这种配合是非常好的。
因此,AAD被反驳,并且DDD通过环丙基卤化物的格氏反应的结果得到证实。 AAD未通过直接测试(溶剂氘化)。 DDD通过该测试,通过DCPH(D)捕获测试,以及涉及反应的动力学分析的测试。
即便如此,简单的DDD也不可能是整个机械故事。它没有考虑部分保留配置的观察结果。从产物分布的动力学分析来看,环丙基格氏反应基的τR的衍生值与典型烷基的相似,接近10-7秒。环丙基的反转和外消旋化的弛豫速率常数为10-11s。使用这些值,平方根法则预测RMgX中配置的保留率约为1%。观察到高达26%。
虽然主要途径似乎是R,但这些考虑指向了一条次要途径X.
2.7.乙烯基卤化物
2.7.1.乙烯基自由基中间体的证据
光学活性的乙烯基溴,4-甲基环己基溴溴甲烷,给予RMgBr外消旋化(58%)和c和s的产物,表明乙烯基自由基中间体:
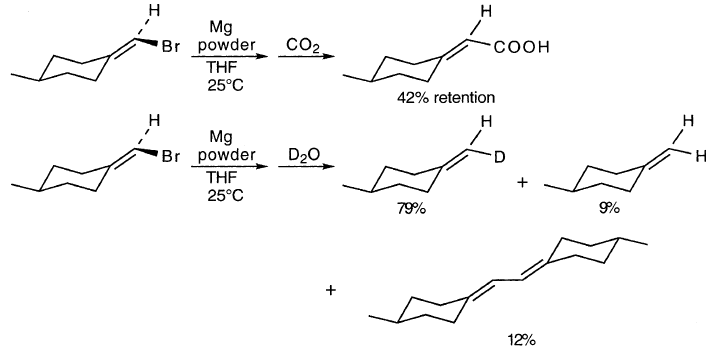
2.7.2. ADD
4-甲基环己基溴甲烷的反应类似于1-甲基-2,2-二苯基环丙基溴的反应(HW,157',161)。因此,Walborsky和同事赞成AAD。
2.7.3. DDD
由于4-甲基环己基溴甲烷和1-甲基-2,2-二苯基环丙基溴的格氏反应得到平行结果,并且由于DDD是后一反应的主要机理,因此它也可能是前一反应的重要机制。 c和s产物的大量产率的形成表明,乙烯基的r-限制寿命蟿R与环丙基和烷基的相似,鈭鈭 7s。乙烯基和环丙基的反转速率常数kQ是相似的,据信,鈭鈭1s鈭鈭对于kQ=1脳1011s鈭鈭蟿R=1脳10鈭鈭,平方根法预测1%保留率远远低于观察到的42%。据报道,在THF中,格氏反应顺式和反式溴代苯乙烯的配置保留更多:
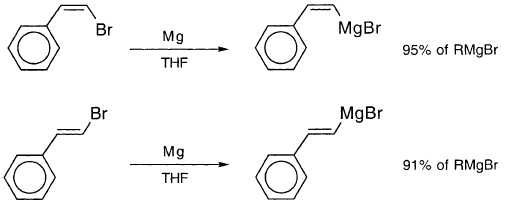
乙烯基系统的结果似乎需要通路X.此外,苯乙烯基案例中的大量保留表明X是主要途径。
2.8.芳基卤化物
2.8.1.芳基自由基中间体的证据
苯基溴在DEE中的格氏反应得到c和s的副产物,清楚地表明苯基自由基中间体:
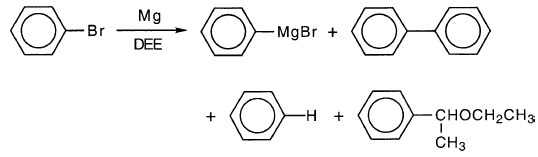
然而,苯基卤化镁的产率可能非常高,对于DEE中的苯基溴通常≥90%,在THF中接近100%。因此,副产物收率通常非常小。指出了自由基中间体,但不一定是反应的主要部分。
尽管邻- (3-丁烯基)苯基在环化反应中具有高反应性,但在37℃时kQ= 4×108 s-1,邻 - (3-丁烯基)苯基卤化物的格氏反应得到低产率的环状产物,特别是在THF中,表6。
表6
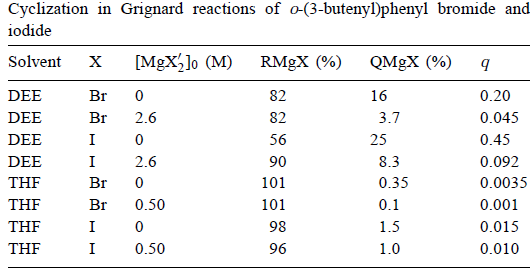
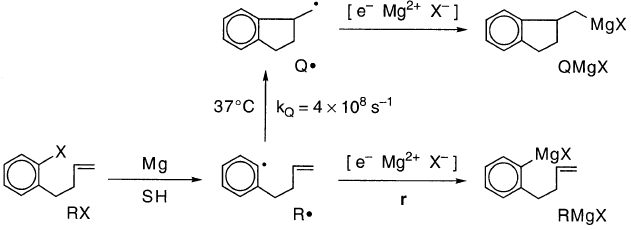
同样,指示芳基是反应某些部分的中间体,但该部分可能是次要的,特别是在THF中。
2.8.2.AAD
根据KR(63),“在诸如苯基,甚至甲基之类的高活性自由基的情况下,它们可以在任何通常的格氏溶剂存在下存活足够长的时间。。。 [c]在很大程度上是荒谬的“。因此,苯基卤化物反应产物中RR的存在表明AAD。
尽管他们很少关注芳基卤化物,但HW(181)认为AAD适用于他们的格氏反应。
2.8.3.DDD
由于环丙基和苯基在其他情况下具有相似的反应性,因此与环丙基卤化物类似,表明DDD用于芳基卤化物的格氏反应。对于烷基,环丙基和乙烯基,τR的值似乎是~10-7 s,因此可以合理地假设类似的值适用于芳基。
τR≈10-7s,DDD严重失败。对于没有添加MgBr2的DEE中的邻- (3-丁烯基)苯基碘,q的观察值为0.45(表6),但是根据平方根法计算值为3.6,τR= 3×10-8 s。对于含有MgCl2的THF中的邻 - (3-丁烯基)苯基溴,q的计算值为3.87,而观察值为0.001。
这种差异表明通路X.在THF中,至少,它可能是主要途径。
2.9.反对DDD的论点的谬误
通过反对DDD来支持AAD,KR和HW忽略了以下两者之间的差异:
1.自由基和表面自由基对的行为,
2.“解决方案”和格里纳德激进派的生命周期,
3.稳态浓度的“溶液”和Grignard自由基,
4.DEE和DEE-d10的kS值(动力学同位素效应)。
2.9.1.自由基与表面自由基对
表面自由基对ZB的性质与自由基对AB(GU,195-202,267-271)的性质显着不同。惰性AB通过逃逸迅速消失,即通过分离并获得从未使原始伙伴再次接触的扩散轨迹。相比之下,ZB永远不会逃避惰性ZB。
这反映了一维和三维扩散之间的根本区别。 ZB是一维的情况,因为唯一重要的方向是x,垂直于表面Z.无论偏差多远,扩散的惰性粒子B将来都会与Z接触。AB是三维情况,因为相对扩散的所有三个分量方向(x,y,z)都是显着的。如果扩散的惰性粒子B不在A附近,那么将来它将与A接触的可能性很小(GU,198-202)。
[MgZ R•]是ZB-扩散分离不会导致逃逸。[R•R•]是AB扩散分离确实导致逃逸。对于曾经接触过的球形分子,逃逸发生的概率是s /(R+ s),其中R是接触半径,s是球体的间距(表面之间的距离)。
对于典型的惰性AB和ZB,图9给出了当t = 0时接触对的存活概率S(t)随时间t的变化。到10-10秒时,超过50%的AB逃逸并变为独立地扩散自由基10-9秒,约85%(图9)。
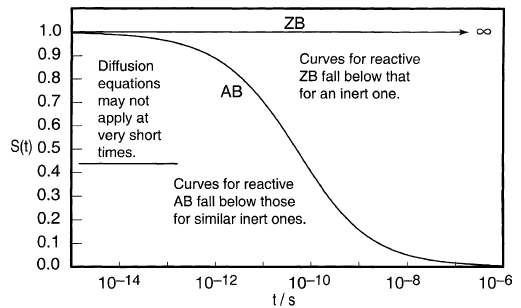
图9
反应性AB通过反应消失并逃逸,因此比惰性AB具有更短的寿命。对于图10中所示的代表性情况,最初接触的50%对在10-11秒消失,其中60%通过逃逸消失,40%通过对反应消失。反应性ZB仅通过反应消失。
所示的情况是[MgZR·]中典型的烷基格氏离子基团。最初接触的50%对消失需要10-8秒以上,90%左右需要10-6秒。[错误:GU(201,图7.11)中类似图形的AB曲线错误地转移到更短的时间。]
如果一对基团[R•R•]在约10-10秒内未发生对反应,那么它可能通过扩散到几个接触半径的分离而逃脱。逃逸的自由基在溶液中独立地扩散。如果原始的自由基对在溶液中均匀地产生,则逃逸的自由基表现得像单独和均匀地产生的那些。它们的寿命很长,从未达到很高的浓度。这允许它们经历(假)一级反应,例如溶剂侵蚀和异构化,并且如果它们具有足够的反应性,则使它们不可能彼此偶联和不成比例。
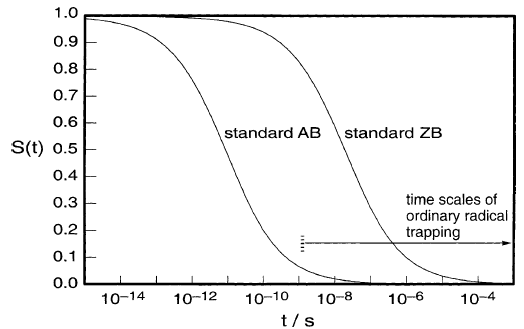
图10
这些是KR和HW的“解”基团的行为,他们认为离开MgZ的格里纳德自由基将表现得相似。因此,KR(63)认为认为苯基或甲基格氏自由基在溶液中能够长时间存活以进行c是“荒谬的”,并且在Walborsky和Aronoff之后,HW(160-161)认为l-甲基-2,如果它们离开MgZ并在溶液中扩散,则2-二苯基环丙基格氏基团必然会经历s或q(开环)。
这两个想法都不正确。[MgZR•]没有逃逸,在MgZ附近扩散的自由基不像“溶液”自由基。在他们的推理中,KR和HW忽略了[(2)和(3)]更短的寿命和更高的格利雅德自由基浓度(接近MgZ)。
2.9.2. “解决方案”与格里纳德激进派的生命周期
简单计算给出了对生命周期限制和烷基“溶液”自由基稳态浓度的保守估计(GU,248)。前面提到的测量和计算为格氏反应提供了类似的信息。寿命τ和烷基“溶液”和Grignard自由基的稳态浓度[R•]。
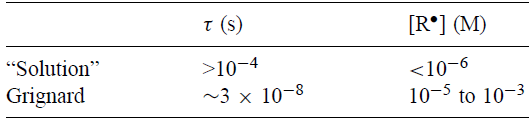
烷基格氏自由基的寿命受到r值的限制,该值至少小于10-4s,即“溶液”自由基寿命的下限。因此,“溶液”自由基将经历与(伪)一级速率常数高达104s-1的反应,这对于相同类型的格氏反应基团是不可用的。速率常数接近105 s-1的反应将主导“溶液”自由基,但对于相同种类的Grignard自由基来说,虽然是显着的(平方根法则),但是很小。
这解释了为什么溶剂侵蚀(kS≈103s-1)在烷基卤化物的格氏反应中不显着。它还解释了为什么5-己烯基(kQ = 4×105s-1)的环化对于“溶液”自由基基本上是完全的,但对于格氏反应基团仅发生3-10%的程度。为了解释这些和类似的差异,没有必要假设Grignard自由基保持吸附在MgZ上。
1-甲基-2,2-二苯基环丙基“溶液”基团R•攻击溶剂或广泛异构化(HW,151-152):
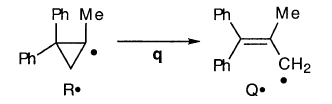
引用Walborsky和Aronoff,HW(162)引用了在DEE中1-甲基-2,2-二苯基环丙基溴的格氏反应中s和q两者的微不足道作为AAD的证据。然而,DDD将这些事实视为R•的短r限制寿命的结果。此外,似乎有比Walborsky和Aronoff更多的思想- 参见第2.9.4节。
2.9.3.“溶液”与Grignard自由基的稳态浓度
Grignard自由基比“溶液”自由基偶联和不成比例的倾向反映了Grignard自由基的较高浓度,通过10-103或更多的因子。对于高浓度(~1 M或更高)的烷基卤化物的格氏反应,在MgZ处,自稳态浓度的自由基浓度接近10-3 M,在溶液中接近数千埃时超过10-5 M(图6)。因此,对于Grignard而言,比率c/ q的比率将是10-103或更高,而不是“溶液”自由基:
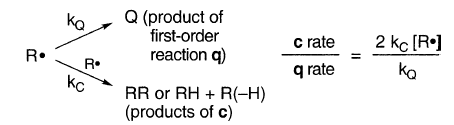
在格氏反应中,当kQ为104s-1或更小时,c将发生近似排除q。这可以通过将MgZ附近区域中的有效[R•](其中c出现)设为10-4M(图6)来计算,因此c rate / q rate =(3×109M-1 s- 1)×(10-4M)/104s-1 = 30。
MgZ附近的[R•]的高值解释了c在Grignard反应中如何显着,而s不是,即使相同类型的“溶液”自由基在10-7M处经历比c更多的s:
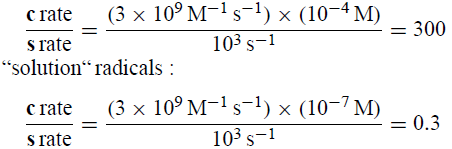
它还解释了c如何在格里纳德反应中与r竞争。应用平方根法则得到c/ r = [(3×105 s-1)(3×10-8s)] 1/2和c= 9%。取决于DEE中RBr的初始浓度和实验方法的流体动力学,观察到的c范围超过0-40%(GU,209)。对于RBr最初为~2.1M并且没有流体动力学控制的反应,观察到的c为~10%,与刚给出的近似计算和适当的DDD计算非常一致(图5)。
引用Walborsky和Aronoff[40],HW(162)认为在DEE中1-甲基-2,2-二苯基环丙基溴(RBr)的Grignard反应中RR的形成意味着中间基团R•不是“在溶液中游离””。 “解决方案”激进分子R•不要耦合- 而是将它们异构化为Q•,它们会联系给QQ。在MgZ附近的短寿命和高浓度的R•是R·Grignard反应产物中的“游离溶液”。
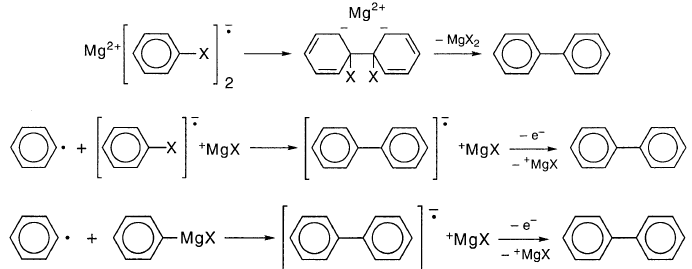
在认为苯基格氏试剂基团不能存活足够长时间以便偶合时,KR(63)忽略了这些自由基可能达到的MgZ附近的高浓度。也就是说,这并不一定意味着在苯基卤化物的格氏反应中形成的少量联苯是苯基自由基偶合的产物。联苯可能是由于苯基卤化物阴离子自由基的偶联或苯基与苯基卤化物阴离子基团或苯基卤化镁的反应造成的:
2.9.4.DEE与DEE-d10的kS值(动力学同位素效应)
对于DEE中的环丙基,通过近亲的测量估计kS = 2×106s-1。由于这是烷基自由基的相应值的~103倍,因此期望通过环丙基格氏试剂基团进行更多的溶剂攻击是合理的。
与此期望相一致,Garst等人。发现近25%的产品用于最初不含MgBr2的DEE中环丙基溴的格氏反应。当存在MgBr 2时,这降低至约6%(GU,214-217)。对于2,2,3,3-四甲基环丙基溴,即使在MgBr2存在下也发现了近70%的s。
在Walborsky和Aronoff之后,HW(162)报道了在DEE-d10中1-甲基-2,2-二苯基环丙基溴的格氏反应中RD的产率为1.4%。忽略了对kS的主要动力学同位素效应的可能性,他们将DEE-d10中RD的产率作为DEE-d0中反应的s程度的量度。得出结论s可以忽略不计,他们声称“缺乏溶剂裂解”表明中间基团R•是“表面结合的”。
在相关反应中,对kS的显着同位素效应以DDD预期的方式影响产物分布(表2和3)。由于忽略了它,Walborsky和Aronoff误判了DEE中1-甲基-2,2-二苯基环丙基溴的格氏反应中s的程度。他们自己的数据显示有~11%s。这是报道的RH(23%)和R(-H)(12%)的收率之间的差异。
没有真正缺乏溶剂裂解。环丙基卤化物的格氏反应中s的数据与DDD一致,并且不支持AAD。
2.10.途径X.
2.10.1.τR对R中结构变化的不敏感性
对于沿DDD的伯烷基格氏基团,τR≈1×10-7s。对于环丙基和乙烯基格氏反应基团,s和c的产物的收率意味着τR的相似值,尽管在其他反应中,环丙基和乙烯基比烷基更具反应性。
为什么τR的值对R•的结构变化不敏感?可以排除r受到控制的可能性。对于τR= 1×10-7s且D= 3×10-5cm2s-1,在扩散到5的分离之前,MgZ处的R•将遭受r的概率仅为0.03。
也许r率受复杂腐蚀中的其他事件控制。在反应过程中将MgZ保持在稳态条件下的过程包括Mg作为Mg2+(可能)的溶解,伴随的溶剂和离子聚集体重组,以及RX的降低,以及R•的还原。也许R•以保持所有进程同步所需的速率降低。
2.10.2.单独DDD
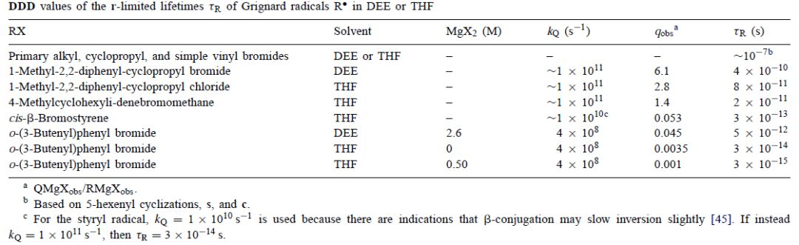
如果DDD是排他性途径,那么寿命τR的值将由平方根法则和kQ和qobs的观察值[QMgXobs/ RMgXobs]给出(表7)。由某些环丙基,乙烯基和芳基格氏反应基团的异构化得到的τR值远小于10-7秒。有几个足够短,10-15到10-12秒,以使扩散方程的适用性和平方根法,可疑。有些足够短,10-15到10-13秒,具有可疑的物理意义。这些结果不支持假设环丙基,乙烯基和芳基卤化物的独特途径DDD。
表7
2.10.3. D7 / X0
纯DDD是不够的。或许另一条途径X与之竞争:

设D7为DDD,τR=1×10-7 s。设X0为路径X,没有基团中间体或异构化。组合是D7 / X0机制。如果假定为D7 / X0,表7的数据提供了足够的信息来将格氏反应分配到D7和X0。由于kQ的所有值都显着大于107s-1,因此沿D7的异构化几乎完全,并且非异构化的格氏试剂RMgX的产率(相对于完全异构化的,即平衡的格氏试剂QMgX)是良好的衡量标准。通过途径X0的反应部分。
即使这样,也可以获得更好的计算结果。使用异构化速率常数kQ的已知值,平方根法则为沿D7形成的产物提供q7的计算值:
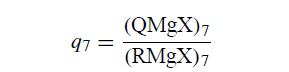
根据qobs[QMgXobs / RMgXobs]]和q7的定义以及质量平衡关系QMgX7+ RMgX7 + RMgXX = 1,可以推导出通过路径X0进行的反应部分的以下表达式:
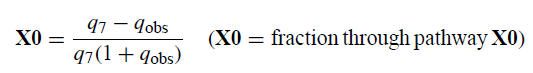
表8给出了进入途径D7和X0的反应的分区。
该结果提供了这样的趋势的定量表达:RX中的缀合增加与途径X的增加部分相关。
表8
2.10.4。D7 / XR
在另一种情况下,基团R1'是沿路径X的中间体,表示为XR。让R的寿命非常短暂,因为它是一对双子座的成员。在D7 / XR中,存在两个中间自由基R'- D7组(蟿R = 1脳10鈭)和XR组(蟿R 1脳10鈭)的子集。如果MgZ与活性位点F“相关”,则如果R'在F处由RX形成,并且如果在形成R'之后该F保持活性,则R'和F将形成成对的对[FR]三维。这一对的行为类似于解决方案中的一对激进对的行为。它的寿命非常短,即使这个寿命主要受到逃逸的限制(图9)。如果没有发生成对反应,那么R'将逃逸并进入在MgZ附近扩散的D7自由基池。除非kQ非常大,否则Geminate反应r(XR)将导致RMgX,在这种情况下将形成一些QMgX:
在D7/ XR情况下,异构化速率常数远大于107s-1的自由基探针将沿着D7平衡并且可以沿XR部分平衡。原则上,这允许D7/ XR在实验上与D7 / X0区分开来。对于D7/ X0,只有异构化的时间尺度τR(τR= 1×10-7s)。如果kQ足够大以至于几乎完全平衡,则进一步增加将几乎不产生额外的异构化。对于D7 / XR,kQ的进一步增加将提供显着更多的异构化。下面描述了尝试测试这种情况的尝试。
2.10.5。有雀斑且均匀反应的MgZ
原则上可以提出异议。如果在雀斑处发生减少,那么MgZ将不会如上述DDD动力学处理中所假设的那样均匀反应。由于DDD处理成功,可能有人认为MgZ不能被雀斑,XR不能成为通路X.实际上,成功的处理只需要系统表现得好像MgZ是均匀反应的。当R•处于MgZ时,存在α概率,它将反应形成RMgX而不是分离。如果表面均匀反应,则相同的值?适用于所有MgZ-R•遭遇。相反,如果表面有雀斑,α的值将随遭遇而变化,但系统的动力学可能表现为MgZ与α的平均值均匀反应。对于足够长寿的自由基和足够高密度的雀斑来说就是这种情况。如果要排除XR,则必须以其他理由为依据。
2.10.6. XR的证据
Garst等人的自由基环化产物收率非常低。为了表明对于芳基卤化物的格氏反应,主要途径是X0:“沿主要反应通道没有芳基中间体”。然而,他们的数据并不排除XR,这可能是由更快的激进探测揭示的。使用假定比邻 - (3-丁烯基)苯基环化更快的探针,Bodineau等。试图探测中间基团R•的寿命很短:
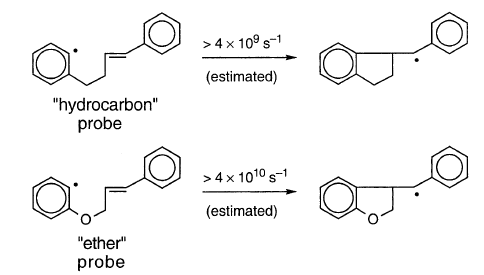
更快的探针确实导致异构化增加。对于室温下在THF中的Mg *(原子团簇)与对应于“烃”基团的芳基溴反应,q的观察值为0.15。对于与Garst等报道的邻- (3-丁烯基)苯基溴[57]和q= 0.0035(两次实验的平均值)的类似实验,这与q = 0形成对比。对于使用普通Mg的相同反应。从更快的“醚”探针的溴化物的格氏反应中发现更多的环化,其中q = 1.6。使用“碳氢化合物”和“以太”探针的kQ的估计最小值,平方根定律和q的观测值给出“碳氢化合物”探针的τR=6×10-12s,τR= 6×10- 11秒为“以太”探测器,值远小于10-7秒。
这些数据表明XR占主导地位。邻- (3-丁烯基)苯基溴的数据排除了D7,并且用更快的探针增加的异构化程度似乎排除了X0。但是,这种解释存在不确定性。
2.10.7。极快探针可能引起并发症
根据快速“碳氢化合物”和“以太”探测器的结果排除X0以支持XR可能是不合理的。可能的并发症笼罩着这幅画
后处理后,苯酚是“醚”基质的格氏反应产物之一。假设它是由RMgBr中C-O的还原性裂解引起的,Bodineau等人。将苯酚计为RMgBr:
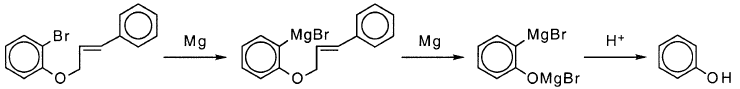
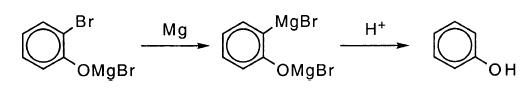
苯酚还有另一种可行的途径。副产物之一是邻溴苯酚,表明在C-O键的初始裂解而不是C-Br键的情况下发生还原。作者没有评论邻溴苯氧化物可能被Mg还原成酚盐的可能性,在这种情况下将苯酚计为RMgBr是错误的:
“醚”探针的另一个可能的并发症涉及氧的作用。醚与Mg2 +配位。涉及底物的配位可能影响格氏反应的过程。
对于“烃”和“醚”基质,还有另一个复杂因素。芳基溴部分不是唯一合理的还原位点。第二个是苯乙烯基团。实际上,“醚”底物的还原为邻溴苯酚提供了合理的途径,这可能是该产品的起源:
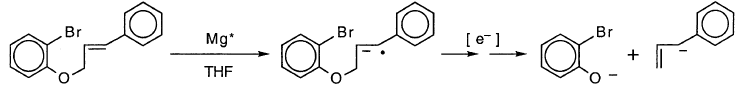
这特别可能是由于“醚”底物的脱溴类似物经历该反应的事实。
苯乙烯基的还原是“烃”和“醚”底物的格利雅反应的合理的第一步。人们可以设想循环产品的后续途径,包括QMgX,它在下面的方案中表示为阴离子:
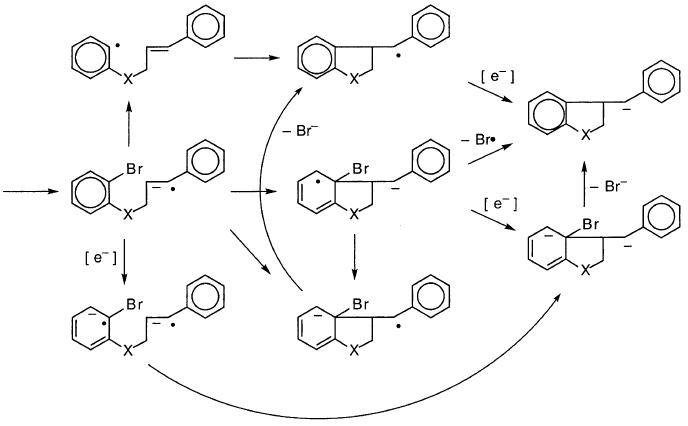
这些考虑使人对“碳氢化合物”和“醚”底物作为格氏试剂形成机制和其他还原机理的探针的有效性产生怀疑。相应的格氏反应中的异构化可能是通过其他过程产生的伪影。
沃尔特的一份报告引发了额外的疑问,沃尔特在DEE中研究了邻烯氧基碘苯的格氏反应。尽管该探针的kQ值很大,但没有检测到异构化的格氏试剂:
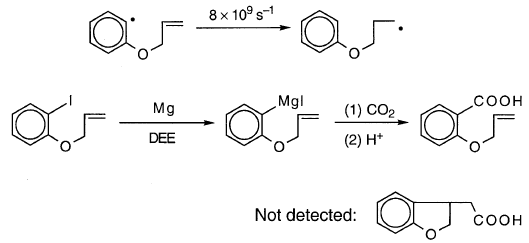
奇怪的是,与Garst等人相比,Walter发现较快的探针o-烯丙氧基碘苯的异构化较少。发现邻 - (3-丁烯基)苯基碘[52]。此外,尽管事实是(1)邻烯丙氧基苯基具有与“碳氢化合物”探针相似的kQ值和(2)Garst等人。发现碘化物的异构化比溴化物更多,DEE中的异构化比THF更多,“碳氢化合物”溴化物/THF的异构化比邻 - (3-丁烯基)苯基碘化物/DEE的异构化更多。
在模糊性得到解决之前,应该谨慎地考虑“碳氢化合物”和“其他”提供的XR证据。
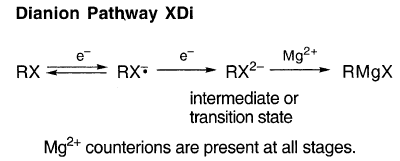
2.10.8. Dianion途径XDi
如果应用XR,为什么X的程度与增加的共轭相关(表8)?这可以通过“ianion”路径XDi更好地解释。在RX转换为RMgX时,两个电子(正式地)从Mg传递到RX。沿XDi传输第二个电子,而R和X仍在一起。如果“渆”电子占据双阴离子中间态或过渡态的轨道,则增加共轭将使其稳定:
2.10.9。溶剂和盐的影响
具有更高的介电常数,THF(7.4)是比DEE(4.3)更极性的溶剂。对于邻- (3-丁烯基)苯基溴和碘化物的格氏反应,异构化在从DEE到THF的过程中降低(表6)。类似地,当通过添加盐(DEE中的MgBr 2,THF中的MgCl2)来增加介质的极性时,异构化降低。通路X的表观程度随着培养基极性的增加而增加(表8)。这与XDi一致吗?
在气相离子对中,较小和较高电荷的阴离子与金属离子的相互作用更强,即它们具有更高的“阳离子亲和力”。具有较高阳离子亲和力的阴离子也具有更强的离子 - 偶极相互作用,包括氢键。
在离子是游离的极性质子溶剂中,更高阳离子亲和力的阴离子更有利于更极性的溶剂,由此它们更强烈地溶剂化。在极性较小的非质子溶剂中,离子是离子对或更高的聚集体,较低阳离子亲和力的阴离子有利于极性较小的溶剂,其中在离子间库仑吸引力和离子溶剂化之间竞争较少。这是溶剂效应(PISE)反转原理的本质。
对于XDi,过渡态阴离子将具有比导致R•的单阴离子过渡态更高的阳离子亲和力。 PISE的天真应用表明更极性的醚应该有利于途径R.这与观察到的相反。
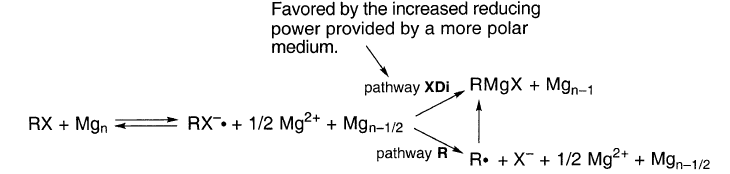
实际上,PISE仅适用于金属离子是观察者的情况,也就是说,当阴离子从反应物变为产物时,它们既不会形成也不会被破坏。在格氏反应和其他金属腐蚀中,金属离子在反应本身中形成。
通过溶剂化和离子聚集,Mg2+的损失及其在介质中的稳定化导致Mg的减少。极性更高的醚THF提供更好的阳离子溶剂化,因此比DEE具有更大的还原能力。类似地,含有盐的醚比无盐醚提供更多的还原能力。
更多的降低功率可以增加来自RX的XDi跃迁状态[RX2-]‡的形成率- 而不会增加RX• - 通过碎裂消失的速率。因此,更多的极性媒体可以支持XDi:
尽管对高极性溶剂中芳基卤化物的电化学还原进行了深入研究,但尚未检测到双阴离子还原。在电化学还原中,通过广泛的离子聚集没有稳定双阴离子过渡态,抗衡阳离子是观察者,并且它们不是金属阳离子。也许这些差异可能使得双阴离子途径在格氏反应中受到青睐,但在相同芳基卤化物(GU,224-225)的电化学还原中则不然。
2.10.10。激进的寿命和扩散程度
对于通路XR,考虑自由基R•的非常短的寿命的影响。他们会从MgZ扩散到多远?
偏移概率χ是一个有用的量度(GU,202)-χ是除了r之外的每个反应中惰性的R•将达到与MgZ的分离s(在经历r并转换为RMgX之前)的概率:
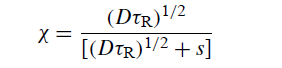
令s1 / 2为χ=1/2的偏移距离。然后s1 / 2由以下关系给出:
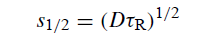
表9给出了τR的各种值的s1/ 2。
对于在THF和THF / MgCl 2中的邻- (3-丁烯基)苯基溴,τR的平方根定律值接近10-14s(表7),对应于s1/ 2 =0.055Å。在这个时间尺度上,几乎没有扩散,并且在由MgZ和溶剂分子限定的“笼子”内,分子的显着运动是伪振荡的。实际上,表7中τR的所有值都是~10-10s或更小,“烃”和“醚”探针的值也是,对应于s1/ 2~5或更小。
如果扩散不重要,那么也许平方根法则不适用。相反,一个简单的一阶定律q =kQτR可能是合适的。在此基础上重新计算τR给出更长的寿命,在10-12至10-10s的范围内,最短的是2×10-12s[邻 - (3-丁烯基)苯基溴/THF / MgCl2]。对于“烃”和“醚”探针中的每一个,相应的值是4×10-11s,对应于s1 / 2 =3.5。即使在此基础上,我们必须得出结论,如果芳基R•是中间体,它在其寿命期间永远不会远离MgZ(表9)
表9

沿着XR路径,R•和X-被创建为溶剂笼,接触,成对对。 R•和X-在如此短的时间尺度上发生了多少分离,这些分离是从观察到的异构化程度得到的?
对于D = 6×1011Å2s-1(两个扩散物种,对于单个扩散物种来说是两倍)和从一阶定律得到的最小τR(2×10-12 s)值,(6DτR)1/ 2 =2.7Å,意味着在R•的寿命期间R•与X-几乎没有分离。如果应用平方根法则,τR的值将更小并且分离将更少。图9还暗示了一个小的分离,从中可以看出,70-90%的A和B接触产生的惰性AB对在10-12到10-11秒之间存活- 其中许多将留在或将要返回联系。
通路XR的假设意味着必须发生大部分观察到的异构化,而R•和X-是溶剂笼中的接触配偶体。这样的R•会不是一个无可争议的激进分子?它的异构化速率常数是否与溶液中“自由”基团的异构化速率常数相同?
2.10.11。Anton-radical RX• -
文献表明,与溶液中的X-相邻的R•可能不会作为未受阻碍的基团,并且其异构化速率常数可能小于溶液中“游离”自由基的那个。西蒙斯提供的证据表明,邻近的碳中心自由基和卤离子至少是微弱的。通过ESR,在低温基质中,他观察到苯基碘的阴离子基团[62-65]。他假设?* - RX•- 是什么片段,不是?* - RX• - ,而且?* - RX•- 可以经历类似于R•的反应:
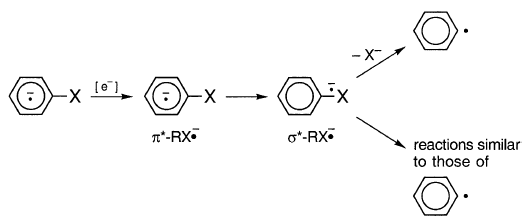
如果,对于芳基格氏反应,异构化和还原的物种是相邻对[R•X−],,如生命时间所示,如果假定通路XR,那么该物种是RX的一个版本。如果在R'和X-之间存在显着的σ键,那么相邻的对实际上是σ∗-RX•-并且基于无阻碍的基团R'的环化率的寿命计算是无效的。 π∗-RX•-可能是环化和减少的物种,在这种情况下,寿命计算也是无效的:
对于[R•X-],σ* -RX• - 和π*- RX• - 的减少,过渡态具有[RX] 2-的组成,加上抗衡离子和溶剂。因此所有这些都是“双阴离子”通路XDi。沿着XDi,可能发生的任何异构化都不是由于未受阻碍的R•的反应。
类似地,Walborsky提出通过“紧密自由基对”[RX•- •MgX +]减少RX是双离子通路XDi。尽管这一提议并未在1973年引起立即批评,但在另一个背景下的类似命题后来也做了。 1991年,基于SRN中的异常和可能由其假设解释的相关反应,Denneys提出芳基卤化物阴离子基团RX• - 可能是一些反应物中的反应物,这些反应已被赋予芳基R• 。 Bunnett严重质疑这一点,但没有严格驳斥该提案,使此事未得到解决。这里的相关性是Denney和Symons的提议是Grignard试剂形成中双阴离子还原途径的意识形态类似物。
2.10.12。三联机制
伯格等人。已经提出了一种机制,其中成对的“三元组”,[R••Mg+ X-]遭受离子对崩溃(ipc)或自由基对崩溃(rpc):
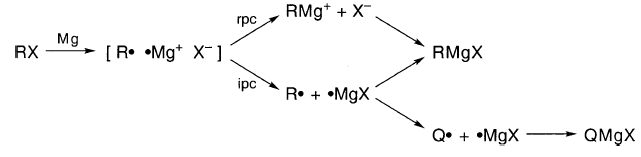
推测Rpc和ipc太快而不能在三联体中形成Q•并且一旦rpc发生就没有进一步的机会。因此,RMgX通过rpc和ipc形成,而QMgX仅通过ipc形成。
三元组机制引入了一层没有证据的复杂性。此外,如前所述,•Mg +和MgX不太可能是中间体。
2.10.13。结论
(1)DDD不能成为芳基卤化物的唯一途径,因为R•的一些隐含寿命τR不切实际地短。必须有一条路径X.
(2)如果芳基卤化物部分通过D7,DDD与τR=1×10-7s反应,部分通过X0反应,其中没有异构化,那么X0的程度有时非常接近100%(表8))。
(3)通路XR,R•是一个寿命极短的中间体,是X0的替代品。
(4)尝试使用非常快速的探针检测XR已经提供了不一致的结果,这些结果受到其他解释的影响。
(5)如果应用通路XR,那么中间自由基R•的寿命将如此之短,以至于许多在成对线[R•X-]中保持在MgZ,直到它们被还原为RMgX。由于基团R•与相邻卤离子的部分键合相互作用[R•X-]实际上是阴离子基团RX的一个版本。因为RX•的减少- 通过双阴离子过渡态[RX2-]‡通过[R•X-]的途径是双阴离子途径XDi。
(6)如果存在类似于α* - RX2-的过渡态,则途径XDi可以解释为什么途径X的程度随着缀合的增加而增加(表8)。
(7)如所观察到的,通过更极性的醚THF提供的增加的还原能力和盐(MgBr 2,MgCl 2)的存在,XDi应该是有利的。
(8)现有证据的平衡有利于XDi作为与R竞争的途径。如果是这样,则XDi对于烷基卤化物可忽略不计,R对于THF中的芳基卤化物可忽略不计。在其他情况下,途径R和XDi都是重要的。
3.“氧化物”层和诱导期
收到后,或暴露在大气中后,表面MgZ涂有“氧化物”层,使其表面无光泽。在格氏反应期间,它经常获得金属光泽,表明已经除去了“氧化物”层。
钝化的“氧化物”层可能是诱导期的原因,诱导期由反应的早期阶段组成。当MgZ完全暴露时,反应可以自由进行。这是“表面清洁”假设。1,2-二溴乙烷通常用作促进剂。虽然有效的行动可能是表面清洁,但事实并非如此。在常规实验室实验中,MgBr 2的初始存在或不存在可能是关键的。因此,在不存在MgBr 2的情况下,在DEE中环丙基溴的格氏反应中存在明显的诱导期,而在其存在下则没有(图1)。
由于BrCH2CH2Br与Mg反应生成MgBr2,因此它可能仅仅因为它产生这种可溶性盐而是有效的促进剂。在促进2,2,3,3-四甲基环丙基溴的格氏反应中,无论MgZ是否在引入RBr之前通过与BrCH2CH2Br反应“蚀刻”,MgBr2都是有效的。当MgBr2不存在时,“蚀刻”无效(GU,257-259)。
这些事实表明了“自催化”假说。极性溶质如MgBr2,也可能是RMgBr,可提高格氏反应的速率。由于它们最初不存在但是随着反应的进行而形成,因此格氏反应是自催化的。它们最初非常缓慢,但随着自诱导期间自催化物质的积累,它们会加速。
无论表面清洁是否是诱导期的原因,在格氏反应过程中MgZ光泽的变化表明它确实发生。理解这些反应需要理解“氧化物”层。
在Abreu的研究中,在超高真空(UHV)装置中暴露出选定的干净单晶Mg晶面。泄漏潜在的反应气体,并通过光谱和衍射方法表征各种表面。通过在高真空室和可以进行普通实验室操作的前室之间转移样品,可以实现液体处理。这种方法可以最大化信息增益。然而,格氏反应通常使用多晶Mg(车削,切屑,棒,粉末等)进行,因此必须研究多晶Mg。 Abreu研究了单晶和多晶Mg;与O2,CO2,H2O和6%HNO3水溶液的表面反应;MgO和Mg(OH)2涂层MgZ与MgBr2和BrCH2CH2Br的相互作用。
早期工作提出了硝酸处理。 “为了去除氧化膜并降低在生产表面时产生的缺陷密度”,Hill等人。通过用6%HNO3水溶液处理来“抛光”MgZ。虽然这种处理可以减少缺陷,但事实证明它不会去除氧化膜。相反,它留下了更厚的电影。这证明在我们的研究中是有价值的,但该方法对于激活镁是无效的。
预计“氧化物”层可能含有MgO,Mg(OH)2,MgCO3,Mg(HCO3)2或它们的某些混合物,但其实际组成是未知的。 X射线光电子能谱(XPS)可以区分O2-和-OH的氧原子以及CO32-和HCO3-的碳原子之间的氧原子。XPS表明,多晶MgZ上的“氧化物”层,无论是“瓶外”还是用稀HNO3水溶液蚀刻,实际上是Mg(OH)2,其外层含有少量Mg(HCO3)2(GU,255)。虽然可以通过烘烤MgZ Mg(OH)2来生成MgO,但未检测到MgO和MgCO3。烘烤后,暴露在大气中几天使表面恢复到其原始状态,主要是Mg(OH)2。
为了确定MgBr2或BrCH2CH2Br是否在从MgZ中去除氧化物或氢氧化物中起直接作用,研究了它们与氧化物或氢氧化物MgZ的相互作用。考虑了两个主要假设。(1)MgBr2通过加速“氧化物”层的溶解来促进表面清洁。(2)BrCH2CH2Br与MgZ的反应发生在“氧化物”壳的下面,并且通过氧化膜中的缺陷或通道渗透而成为可能。
通过对三种类型的氧化物/氢氧化物表面的反应性的比较研究,可以获得该问题的部分分辨率:(i)明确定义的Mg(00 0 1)-MgO(100)单晶表面,(ii)空气暴露的多晶Mg表面,和(iii)水溶性酸蚀刻的多晶Mg表面。良好有序的Mg(00 0 1) - MgO(1 0 0)表面含有最少的缺陷,并且已经显示它对CO2不起反应,当氧化物是氧化物时,酸碱反应进行得相当快。混乱。在
相比之下,多晶样品覆盖有主要由Mg(OH)2组成的层。这种薄膜是无序的,并且存在大量的通道缺陷。他们被发现具有相当的反应性。水溶性酸蚀刻的Mg上的氢氧化物覆盖层的厚度是未处理(未蚀刻)表面上的氢氧化物覆盖层的两倍多。这是一个相关的区别,因为即使通道缺陷仍然很多,增加的厚度也可能强制朝向金属基板的更曲折的路径;如果这种机制是限速的话,那么迟钝的反应率将是一个后果。
通过将Mg(0001)暴露于低剂量的高纯度O2,在UHV室中制备Mg(0001)-MgO(100)表面。从图11所示的低能电子衍射(LEED)图案可以看出表面氧化物的长程有序。该图中包括MgO adlayer的实空间结构。基于俄歇电子能谱(AES)测量,估计MgO膜的厚度为18埃。该表面和促进剂之间的反应在惰性气体(Ar)气氛下的前室中进行。
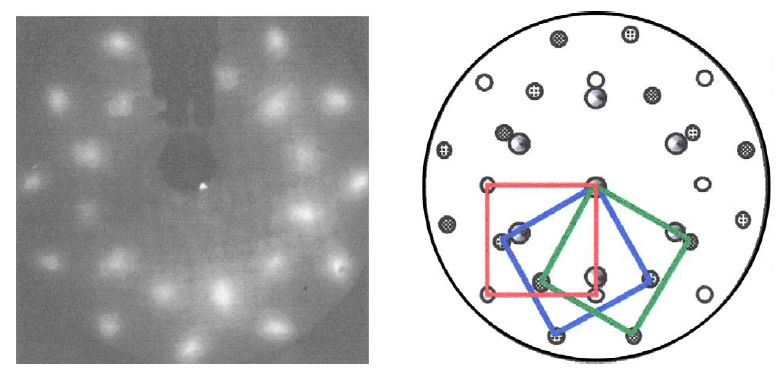
多晶材料不需要UHV样品制备方案。因此,尽管反应也在Ar环境中进行,但是将样品从反应容器转移到X射线光电子能谱仪进行表面表征使得它们短暂地暴露于大气O 2和H 2 O.根据XPS测量,估计未处理样品的Mg(OH)2膜的厚度为16埃;对于用6%HNO3蚀刻的样品,覆盖层厚度大于36埃[5]。
图12显示了在(图12(a))之前和在醚状MgBr2中浸渍3分钟后的Mg(00 0 1)-MgO(100)表面的AES光谱(图12(b))和BrCH2CH2Br(图12(c))解决方案。
对于未反应的MgO(100),仅观察到两个信号,一个用于Mg,另一个用于O.
暴露于MgBr2后,出现了两个新峰;一个在100eV,一个是Br的特征,另一个在270eV是由C引起的.B1的存在很可能是由于固体MgBr2即使在用纯DEE多次冲洗后也没有被除去。另一方面,C的存在可能是残留的BrCH2CH2Br金属Mg之间反应的结果。BrCH2CH2Br是制备醚MgBr2的原料。
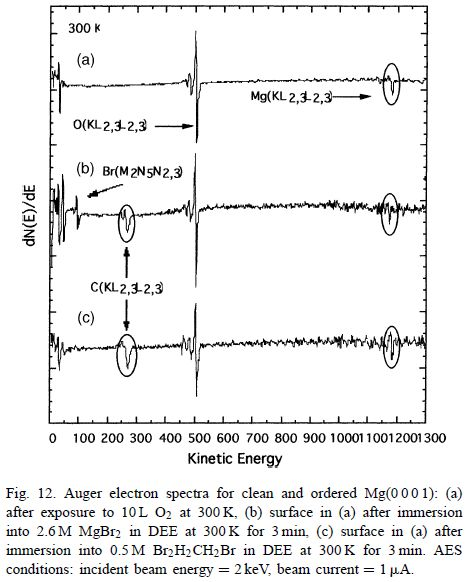
图12
如图12(c)所示,暴露于BrCH2CH2Br的Mg(0001)-MgO(100)的AES光谱表明不存在表面Br。然而,可以注意到O峰的轻微减小,伴随着C和Mg峰强度的轻微增加。
图12(b)和(c)中的数据表明,对于良好有序的Mg(0001)-MgO(100)表面,BrCH2 CH 2 Br仅具有轻微的反应性,而MgBr2是完全惰性的。然而,不能确定Mg-BrCH2CH2Br反应是否先于氧化物的溶解,或者BrCH2CH2Br是否通过(稀疏)沟道缺陷简单地穿过膜。
图12(b)和(c)中的数据表明,对于良好有序的Mg(0001)-MgO(100)表面,BrCH2CH2Br仅具有轻微的反应性,而MgBr2是完全惰性的。然而,不能确定Mg-BrCH2CH2Br反应是否先于氧化物的溶解,或者BrCH2CH2Br是否通过(稀疏)沟道缺陷简单地穿过膜。
MgO溶解的不可信性和O信号保持很大的事实表明了“挖洞”情景。通过实验提供对该视图的支持,其中曝光时间从3分钟增加到30分钟。根据AES光谱,结果如图13所示;这些与图12中的那些完全相同。暗示反应是缓慢的,因为促进剂需要穿过通道缺陷,在良好有序的MgO(100)层中,这些缺陷很少。
关于通道缺陷作用的关键见解可以从多晶Mg与醚BrCH2CH2Br的反应性的比较研究中获得。图14显示了在各种暴露时间浸入BrCH2CH2Br中之前和之后未处理的Mg屑的XPS光谱。如前所述,这里的“氧化物”层主要是Mg(OH)2。
最重要的趋势是,随着反应的进行,O 1s信号(531eV)衰减,而Br 3d信号(70eV)增加。为了确定O 1s峰的衰减是否是由于实际的氢氧化物去除或简单形成不溶性MgBr 2沉淀物,更精细地仔细检查Mg 2p和O 1s峰。图15显示了作为反应时间函数的O 1s峰强度。
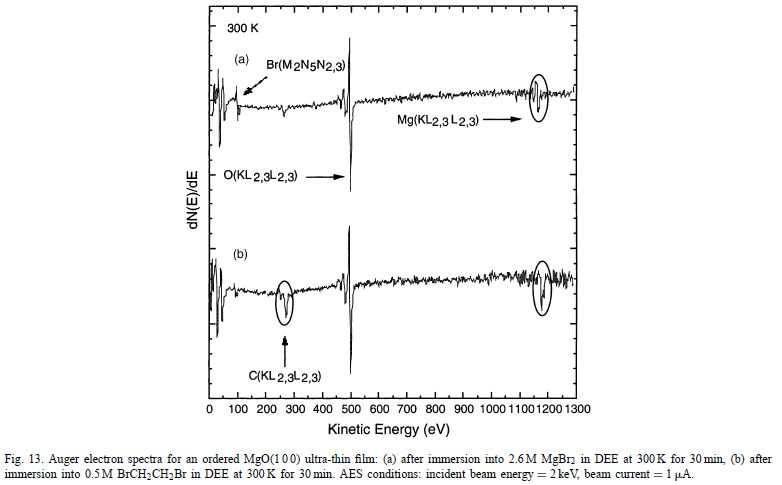
图13
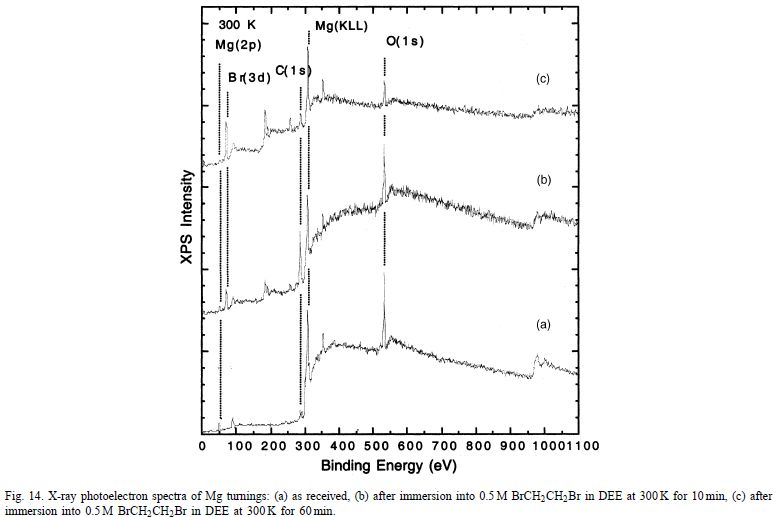
图14
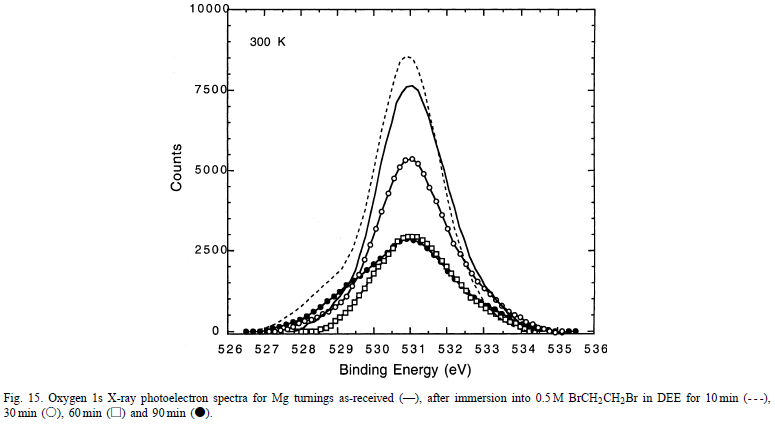
图15
很明显,随着反应时间从10分钟增加到60分钟,表面氧气的量减少。因此证明了暴露于BrCH 2 CH 2 Br后去除原始氢氧化物壳。当反应后的样品在运输到XPS分析室时与大气O 2和H2 O接触时,O表面覆盖率在较长反应时间变得恒定的事实很可能与氢氧化物膜的再生有关。
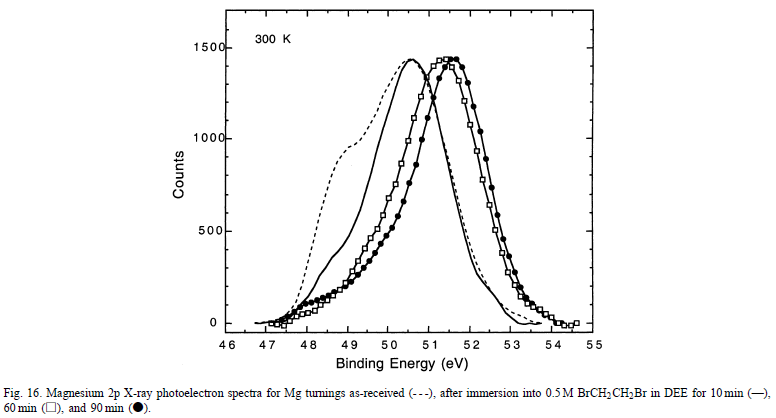
图16
图16给出了Mg2p XPS光谱作为曝光时间的函数。
对于未反应的表面,较低结合能(EB)的肩部是金属镁的氢氧根减弱峰; 50.5 eV的主峰是由羟基化的Mg引起的。随着反应时间的增加,观察到两个主要变化:(i)较低EB值的肩部消失,和(ii)50.5eV的峰值在52eV时转变为较高的EB。与参考化合物的光谱比较表明,52eV处的峰是由于存在块状MgBr 2。这些结果提供了证据,即尽管存在(无序的)Mg(OH)2薄膜,但在BrCH2CH2Br和Mg之间发生了反应。尽管如此,通过底层反应引起的促进剂诱导的化学溶解或物理剥离除去覆盖层的机理仍未解决。
当上述结果与酸蚀刻的镁屑相结合时,可以获得更明确的界面过程视图。图17显示浸渍在BrCH 2 CH 2 Br中10,30和90分钟后酸蚀样品的O1s XPS光谱。
与图15中所示的数据形成鲜明对比,未观察到峰强度的变化。这只能表明没有发生反应。显然,虽然氢氧化物覆盖层中仍然存在通道缺陷,但后者已变得太厚。因此,BrCH2CH2Br通过薄膜所采取的路径变得过度曲折并且仅用于严重阻碍反应。

图17
从上述结果可以得出的结论是Mg(OH)2机械钝化MgZ。BrCH2CH2Br进入MgZ的界面机制是通过沟道缺陷渗漏。当底层反应从金属基材上剥离时,氢氧化物外壳剥落。
有证据表明格氏反应可导致从MgZ释放少量金属Mg。因此,在金刚烷基溴化物的反应中,MgZ涂覆有黑色沉积物,该沉积物可能主要由二金刚烷基组成,但似乎在距离MgZ一定距离处含有金属Mg。作者将远处的Mg解释为•MgBr的歧化产物。
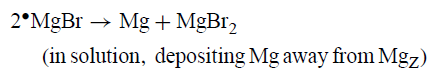
远距离Mg由Mg(OH)2的腐蚀性底切产生更合理的结果。底切不太可能将MgZ与Mg(OH)2完全分离。剥落的材料可能由小块Mg-Mg(OH)2组成,其中一些金属Mg仍然附着在Mg(OH)2层的残留物上(GU,256,图7.37)。如所发现的那样,搅拌或对流将使这些碎片远离本体反应物MgZ。如果碎片足够细,它们也可以使双金刚烷沉积物呈现黑色。BrCH2CH2Br既是Grignard反应促进剂又是模型底物。据推测,格氏试剂BrMgCH2CH2MgBr不是最终产物的原因是中间体如BrCH2CH2MgBr分解成CH2 = CH2和MgBr2。
正如对于BrCH2CH2Br一样,在纯DEE中,Mg(OH)2的覆盖层将机械地阻止任何RX进入MgZ。这足以说明在最初不存在MgX2时观察到的诱导期。
事实上,在DEE中最初存在MgBr2消除了诱导期,如图1所示,仍然需要解释。三种似是而非的行为反映了MgBr2-DEE的极性高于纯DEE。(1)通过润湿和穿透无序的Mg(OH)2层,可能扩大其缺陷通道,MgBr2-DEE可以增强RX向MgZ的扩散。(2)通过溶解Mg(OH)2层,MgBr2-DEE可以清洁MgZ。(3)通过提高MgZ的还原能力,MgBr2-DEE可以促进电子向RX的转移。这些可能性并不相互排斥。
4. 结论
对于烷基卤化物,Mg与RX反应中的格氏试剂形成通过在表面MgZ附近的溶液中扩散的中间基团R•(路径R/ DDD)。 R•的寿命τR受MgZ下降到RMgX的限制,接近10-7s。
对于环丙基或乙烯基卤化物,具有类似τR值的途径R / DDD描述了部分反应。另一部分是路径X,没有中间R•或者,如果存在,它具有极短的寿命。对于具有延长的缀合的乙烯基卤化物,途径X可以占主导地位。
烷基,环丙基和乙烯基的τR值的相似性可能是复杂金属腐蚀过程的所有步骤必须保持同步的结果。除了电子转移到R•之外,这些步骤还包括从MgZ到溶液中Mg2+的损失,伴随的溶剂重组和可能存在的卤化物离子以容纳Mg2+,电子转移到RX,以及可能在阳极和阳极之间传导离子。 MgZ上的阴极部位。
对于THF中的芳基卤化物,途径X主导近R排斥。在DEE中,沿路径R存在更多反应,但其程度仍然很小。卤化镁,DEE中的MgBr2或THF中的MgCl2的存在增加了途径X的重要性。
共轭程度和通路X之间的相关性表明它是二价阴离子通路XDi,即第二电子被传递到RX而其组分R和X仍然在一起的通路。XDi的过渡态具有[RX2-]‡加上抗衡离子和溶剂的组成。
“氧化物”层比赋予多晶Mg,其暗淡的外观主要由Mg(OH)2组成,厚度约为16埃。它的外层含有少量HCO3-。没有可检测量的O2- 或CO 32 - 存在。
氢氧化物层是机械钝化的。对于纯DEE中的RX,初始反应很慢,因为RX必须通过覆盖层中的缺陷通道到达MgZ。一旦RX或诸如BrCH2CH2Br的助催化剂达到MgZ,Grignard反应就会使Mg(OH)2层底切,然后剥落,留下MgZ暴露。这发生在诱导期,之后反应迅速进行。
在DEE中,MgBr2的初始存在可以消除诱导期。这可以通过MgBr2-DEE在润湿,渗透和溶胀Mg(OH)2覆盖层中的一种或多种作用;解散它;通过稳定溶液中的Mg2+来提高MgZ的还原能力。
致谢
J.F.G.感谢格鲁吉亚大学的图书馆和互联网特权,特别是办公室和实验室空间,用品和办公设备的化学系。这里描述的特高压表面工作是基于J. B. Abreu博士的研究。MPS感谢Robert A. Welch基金会的部分支持。
原文标题:《Grignard reagent formation》
原文出处:GARST, John F, SORIAGA, et al. Grignard reagent formation[J].Coordination Chemistry Reviews, 2004, 248(7):623-652.
涉及格氏试剂的形成


目前评论: