- A+
强调
1.在假紫快速放热环化期间于微反应器中捕获中间体。
2.使用惰性流体载体全氟己烷获得段塞流,所述惰性流体载体全氟己烷是惰性的,并且与反应物/产物不相混溶。
3.动力学在等温条件下研究,允许可靠的反应网络。
4.开发的动力学模型与实验数据一致。
摘要:使用常规离线技术难以研究快速放热反应的机理和动力学。本文讨论了使用团状流(微批次)微结构反应器(MSR)进行高放热快速反应模型(假紫环化)的实验研究。使用交叉连接获得团状流,并确定合适的与参与反应的物种不相容的惰性流体载体(全氟己烷)。MSR为连续的团状流提供了短的反应时间,允许捕获反应中间体并监测动力学。小的反应体积可通过转移反应热来抑制反应温度升高。在等温条件下研究了该反应动力学,并提出了更可靠的反应网络。最后,实现了段塞内的混合以及其对所得结果的影响。该方法具有通用性,可应用于各种快速放热均相反应。
1.前言
在快速放热反应的情况下,浓度分布的研究通常依赖于连续在线监测(例如停流技术[1])或离线分析[2]的测量。在前一种情况下存在两个困难:整合有效冷却以抑制温度升高以及分离中间体以进行鉴定。因此,只有通过离线方法结合样品淬火(冷冻/化学)才能捕获中间体,提供有关反应机理的信息。离线分析的猝灭要么是通过向连续流[3]中添加试剂(称为淬火流技术)完成的,要么是通过向收集样品的容器中添加试剂完成的。
可以使用微结构反应器(MSR)来监测在完全转化之前的毫秒反应时间内的反应物、中间体以及产物的浓度。这种被定义为微型化系统,其具有至少一个能提供高的传质传热速率的亚毫米级范围的尺寸。它们已成功用于快速高放热反应的连续化操作和涉及有毒及爆炸性化学品的转化,如硝化[4]、氢化[5,6]、聚合[7,8]、氧化[9]、卤化[10]、烷基化[11]、四唑合成[3]和重氮甲烷反应[12](更多文献,参见[13-19])。
将反应物快速混合至分子规模和具有明确的停留时间是选择MSR的重要准则。然而,如果没有复杂的MSR几何结构和非常高的压降,这将难以实现快速反应足够快的混合要求(混合时间<0.1s)[20]。即使是在高速流动下,微毛细管反应器中的流动由于尺寸小而呈层状,导致出现抛物线速度分布以及不均匀的停留时间分布。其解决方案便是团状流MSR,其将两种反应物结合在由惰性流体携带的分散的微升段塞(液滴)中。每个分离的段塞充当一个微批反应器。该概念已用于研究酶促反应的反应动力学[21]、苯乙烯的溴化[22]、乙酸对硝基苯酯的水解[23]、高通量催化剂的筛选[24]。在这些研究中报道的反应都具有中等焓和相对慢的动力学。
将假紫罗兰酮(PI)环化成β-紫罗兰酮被认为是模型反应,其代表在精细化学工业中进行的环化反应的类别。该反应在商业上是重要的,因为β-紫罗兰酮被用于合成维生素A,也可用作香料。但是由于该反应的高放热性,反应通常以半分批模式进行:将PI加入由不混溶的有机溶剂(甲苯)和浓H2SO4组成的两相体系中发生反应。在反应完成后,进行水解反应,通过用水稀释H2SO4将产物转移到有机相中。该步骤也是高度放热的。因此,整个过程包括两个放热阶段:PI在浓硫酸中的反应和水解时在H2SO4稀释过程中将产物萃取到有机相中。为了控制在该两个阶段期间的热释放量,反应器中的温度需保持在约0℃,这使得反应速率变低、反应时间变长。使用该方法发现β-紫罗兰酮的选择性约为80%[25]。最近使用1-硝基丙烷作为可与有机物和H2SO4混溶的溶剂实现了高达99%的β-紫罗兰酮选择性,并且消除了双相系统中存在的传质阻力[25]。
在PI到β-紫罗兰酮的整体转化中,主要分为两个反应步骤:先将PI环化成α-紫罗兰酮(步骤I),然后将α-紫罗兰酮异构化为β-紫罗兰酮(步骤II)。我们发现步骤I很快,而步骤II的速度适中。然而,反应的高放热性使得以分批模式研究反应的动力学和机理变得复杂。快速步骤I特别需要在完全转化之前在毫秒反应时间内监测反应物、中间体和产物的浓度。此外,反应的高放热性(反应热=-120 kJ/mol PI)需要有效的散热以避免温度升高的影响。
本文描述了使用团状流微结构反应器进行快速高放热PI环化反应机理的实验研究。将均相反应混合物封入段塞中,使用与参与反应的物种不混溶的载液进行移动。在等温条件下研究了该反应的动力学,并提出了更可靠的反应网络。开发的动力学模型与实验数据一致。最后,实现了反应物在段塞内的混合及其对所得结果的影响。
2.实验
这项工作的主要部分是研究快速步骤I的动力学。然而,为了建立PI到β-紫罗兰酮的整体转化网络,进行步骤II的互补实验(α-紫罗兰酮异构化成β-紫罗兰酮),步骤II反应在批处理反应器中进行。在以下小节中讨论了两种实验装置。
2.1 MSR实验
连续MSR装置由连接到螺旋形毛细管MSR下游的PTFE交叉连接组成,其浸入连接到恒温器的恒温水浴中,恒温器可在243-473K的温度范围内供应热流体。双注射泵(kdScientific Legato 200)用于将两种流体(PI和H2SO4在1-硝基丙烷中的溶液)引入交叉连接的侧通道中。使用单个注射泵(kdScientific KDS 100)引入载体相全氟己烷。将交叉连接前的0.5mm毛细管浸入与交叉连接相同的水浴中。为了能够将流体冷却到所需温度,从MSR的特征冷却时间研究这些毛细管上游的长度,即设定特征冷却时间等于上游停留时间。对于停留时间的变动,下游毛细管的长度在10到100 cm之间变化。
在进行MSR实验之前,在实验室烧瓶中研究了1-硝基丙烷和紫罗兰酮在全氟己烷中的传质。进行两组分配实验:在全氟己烷中的纯1-硝基丙烷和在1-硝基丙烷-全氟己烷双相体系中的紫罗兰酮。发现1-硝基丙烷在全氟己烷中的溶解度在0℃时为37 g/L,在25℃时为57 g/L。然而,我们还发现在全氟己烷中的紫罗兰酮转移可忽略不计(远小于1%)。因此,用1-硝基丙烷饱和的全氟己烷用于实验。
通过在实验中改变不同的操作条件以建立两相流中的流动状态,并且发现0.4 mL/min的总反应流速和1mL/min的饱和全氟己烷条件下,不论反应温度多少均可产生明显的团状流。我们将出口反应混合物收集在含有Na2CO3水溶液(10%w/w)的烧瓶中。然后将烧瓶浸入冰浴中,磁力搅拌所得混合物。因此,本反应通过加入水溶液快速萃取/中和H2SO4来淬灭。最终形成三相:含有1-硝基丙烷的紫罗兰酮、稀硫酸和全氟己烷。分离有机相(1-硝基丙烷+紫罗兰酮),并用无水Na2CO3干燥。使用气相色谱(PerkinElmer AutoSystem XL装置,其配备有30m Stabilwax(Crossbond Carbowax PEG,Restek,USA)0.32 mm毛细管柱,具有0.25l涂层)分析样品。将实验重复三次以检查重现性,并考虑平均浓度用于进一步分析。
2.2 分批实验
使用配备有搅拌器以混合反应组分的分批夹套反应器。将反应器连接到恒温器,该恒温器可以在243-473K的温度范围内供应热流体。该装置可以以批量或半批量模式操作,其中手动控制的计量系统固定在反应器的顶部。通过具有约±1℃变化的传感器监测反应器内的温度。使用注射器取出样品并使用气相色谱分析。
将已知量的α-紫罗兰酮注入到H2SO4和NP(1-硝基丙烷)的混合物中,并以规则的时间间隔取出样品。将水加入到样品中,形成H2SO4水溶液(60wt%)和有机相的两相混合物。再分离有机相,用NaHCO3溶液洗涤以中和痕量的H2SO4,再用无水Na2CO3干燥并通过GC分析。
2.3 化学品和操作条件
紫罗兰酮(PI,α-紫罗兰酮和β-紫罗兰酮)的纯度大于90%,而1-硝基丙烷和H2SO4的纯度为分别为大于等于98%和96%,目前本工作使用的操作条件列于表1中。
表1 本工作中使用的操作条件

3.结果与讨论
3.1 MSR中的团状流
在这里,我们提出了在团状流MSR中快速且高度放热的PI环化的研究结果[26,27]。为了研究PI环化的整体动力学和机理,应该除去升高温度且影响总反应速率的反应热,以提供几乎等温的环境,要求可以有效冷却的小反应体积。

图1 用于监测PI环化的毫秒动力学的MSR系统
(a)MSR系统的图示(b)内径为0.5mm的团状流的单色快照PTFE毛细管
当选择MSR的材料和几何构型以及惰性载体相以形成反应混合物的分散段塞时,使用在反应中起不同作用的各种化学品向我们提出了严峻的挑战。我们发现具有三个入口和一个出口的特氟隆交叉连接适合于产生规则的团状流。将溶解在1-硝基丙烷中的两种反应物PI和H2SO4引入至两个相对的入口,同时通过第三个入口引入不混溶的惰性载体流体全氟己烷,最终在恒温PTFE毛细管MSR中产生了段塞流(见图1)。尽管两相都是有机相,但在PTFE毛细管中建立了明确的团状流。将几微升的团块嵌入到连续载体相中,形成了厚度为几微米的壁膜。由于在段塞内的流体和连续相的壁膜之间的剪切作用引起了内部循环,确保了段塞内反应物的混合,如图1所示。因此可以将该段塞称为微批反应器。通过使用不同长度但相同内径为0.5mm的毛细管在恒定流速下保持相同的流动特性,反应时间在0.84至3.32秒之间变化。这个狭窄的操作窗口来自于:(1)形成团状流的流速;(2)在恒温浴和淬火烧瓶之间建立连接所需的最小毛细管长度。
3.2分析
初始PI、批次实验(反应时间,tr≥120 s)和MSR实验(反应时间,tr<4s)的气相色谱图如图2所示。除了PI(2个异构体)和α-,β-紫罗兰酮的峰外,在MSR实验中还检测到三个额外的峰(1、2、3峰),而在批次实验中不存在此三个峰。
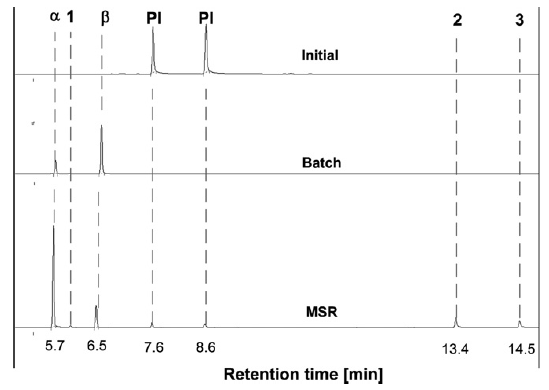
图2 PI环化:初始混合物、批次实验和MSR实验获得的典型色谱图
气相色谱和质谱联用(GC-MS)用于鉴定未知物。发现未知1的分子量为192 g/mol,与α-紫罗兰酮和β-紫罗兰酮的分子量相同,其结构分析显示其为γ-紫罗兰酮。未知2和3的MS指纹是相同的,证实了异构体的假设。通过MS测量得到的分子量为210 g/mol,其高于紫罗兰酮的分子量。分离出的化合物的进一步NMR分析表明它是9,10-双键水合的10-羟基衍生物(6,10-二甲基十一碳-3,5-二酮-10-羟基-2-酮)。值得提醒的是,本实验是通过用H2O淬灭反应混合物来制备分析的样品。
3.3 步骤II的动力学
假设所有步骤都是一级反应,将一组三个质量平衡方程(α-紫罗兰酮、中间物质、β-紫罗兰酮)拟合到实验数据中(图3)。该模型显示ki2≈25ki1,并且速率常数ki1近似等于ki3。它同时还预测了非常低的中间物种浓度,这在采样的样品中几乎不被GC检测到。可得到步骤II的活化能和频率因子分别为65 kJ/mol和5.4×1010 s -1。

图3 步骤II:在间歇反应器中的α-至β-紫罗兰酮异构化:实验数据(点)和模型结果(线)用初始浓度的紫罗兰酮将浓度标准化
在MSR实验中的α-至β-紫罗兰酮异构化期间,仅观察到γ-紫罗兰酮(图4),发现其随停留时间和/或反应温度的增加而降低。这表明γ-紫罗兰酮来源于步骤II中涉及的中间体。这还表明在异构化过程中没有形成PI。
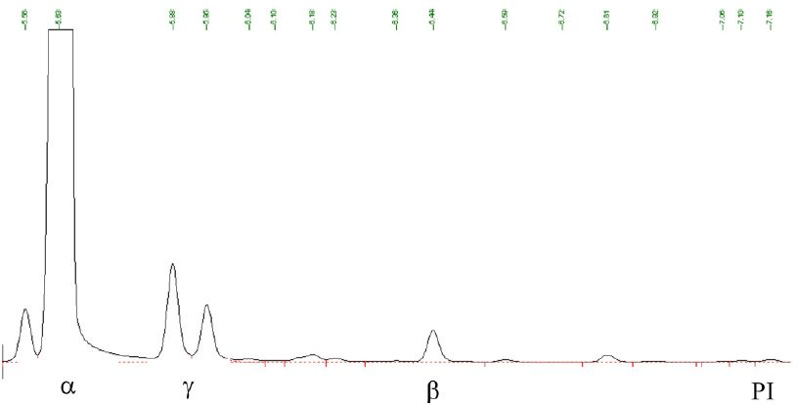
图4 在MSR中进行的异构化实验的典型色谱图显示存在γ-紫罗兰酮
3.4 整体反应网络
从分批实验中了解到,α-至β-紫罗兰酮异构化的步骤II涉及反应中间体(参见图3)。在PI环化的MSR实验中,除了PI(2个异构体)和α-,β-紫罗兰酮的峰之外,还检测到另外三个未知峰1,2,3(参见图2)。由于在第一次分析之前与常规批次实验(tr≥120s)相比,MSR中的反应时间非常短,因此捕获这些中间体成为了可能。如上所述,在α-紫罗兰酮和β-紫罗兰酮之间观察到的峰属于γ-紫罗兰酮,而在较高保留时间的两个额外峰是9,10-双键水合的10-羟基衍生物的异构体(6,10-二甲基十一碳-3,5-二酮-10-羟基-2-酮)。发现10-羟基衍生物和γ-紫罗兰酮的浓度随着反应时间和/或温度的增加而降低,这表明这些化合物分别来自步骤I和步骤II的中间体。从获得的结果可以得到如方案1中所示提出的反应网络。
PI(1)在H2SO4存在的条件下被质子化为(2),形成碳正离子(3)和硫酸氢根阴离子的离子对。由于硫酸氢盐是非常亲核的,因此可以形成磺酸酯和环状碳正离子(6)。在猝灭期间通过H2O分解磺基酯得到10-羟基衍生物(5)。为了完成(6)中键的形成,应该消除三个相邻氢原子中的一个。从位置5,1或从位置6处所示的甲基除去氢将分别得到α-紫罗兰酮(10)、β-紫罗兰酮(12)和γ-紫罗兰酮(8)。β-紫罗兰酮(12)是热力学最稳定的,因此(9)和(7)在酸存在的条件下经过(6)转化为(11)。这种反应机制与Semenovskii等[28,29] (见方案2)先前提出的反应机制相矛盾,在他们的方案中,约90%的PI首先转化为α-紫罗兰酮,然后连续转化为β-紫罗兰酮,而平行路径中的10%PI直接转化为β-紫罗兰酮。本次研究与Semenovskii等人[28,29]的工作还有很多不同。目前的工作是在后一种情况下分离中间体并单独处理。
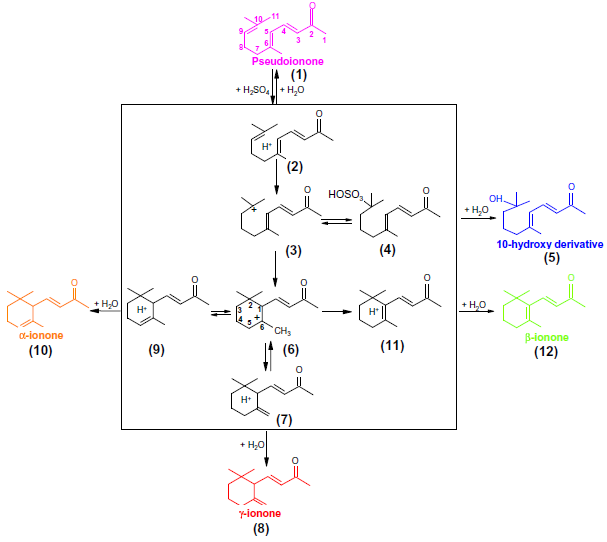
方案1 建议的反应机制,在淬灭样品的GC分析中检测有色组分

方案2 由Semenovskii等[29]提出的PI环化和异构化机理
为了获得PI环化的动力学参数,考虑到可测量化合物浓度的监测。我们假设从(2)到(3)的反应非常快,即中间体(2)的浓度≈0将导致图5中所示的反应方案。进一步假设在(3)和(4)之间以及(6)和(7)之间瞬时平衡。这表示测得的10-羟基衍生物(5)和γ-紫罗兰酮(8)分别对应于(3 + 4)和(6 + 7)。
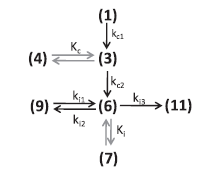
图5 动力学模型开发的简化反应方案
我们开发了质量平衡方程,将所有步骤作为一级反应,由Kc和Ki表示瞬时平衡,如图5所示。为了研究可测量化合物的反应动力学,建立了“真实”和“观察”速率常数之间的关系(详细模型见附录A):
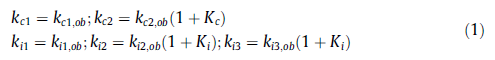
使用商业软件Berkeley Madonna(公差= 0.001)将该模型拟合到MSR实验数据,如图6所示。图中显示的数据点是三次连续测量的平均浓度,标准偏差约为5%。由图可以看出,所有配件(线)与实验数据(点)非常一致。值得一提的是,初始拟合是针对常数kc1,ob和kc2,ob进行的,然后是剩余常数。为了确定所有步骤的表观活化能,从模型获得的观察到的速率常数以Arrhenius形式绘制,如图7所示。
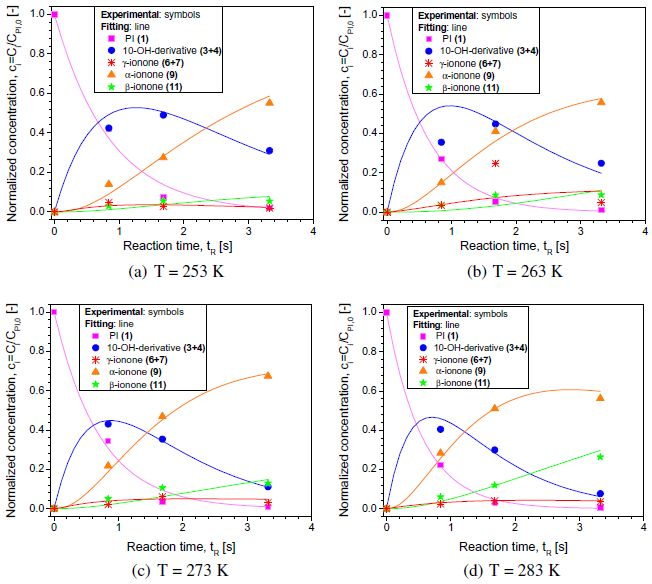
图6 实验(点)和建模(线)浓度分布(T = 273K),用初始PI浓度标准化浓度

图7 用一级动力学模型得到的观察到的反应速率常数的Arrhenius图
在微批反应器中发挥混合作用非常重要。常用的方法是基于光学测量或基于Villermaux的反应。由于在如此低规模下浓度的分辨率,光学方法具有局限性,而Villermaux的反应适用于特定的系统(即需要调节pH),并且不能应用于本工作中选择的化学系统。因此,Tanthapanichakoon等人[30]进行的一项成熟的研究中提到了这方面的内容。他们进行了流体动力学计算(CFD)模拟,以研究微批反应器(也称为微流体段塞)中的混合,并使用Villermaux-uhman反应进行进一步的验证[31]。这是基于改进的Peclet数,Pe*=us(ds)2/lD,其中us是段塞速度,ds是段塞直径,等于通道的横截面尺寸,l是段塞长度,D是试剂的扩散率,其定义用于确定混合是扩散占主导地位还是对流占主导地位。针对两种类型的反应混合物初始排列提出了两种关系:
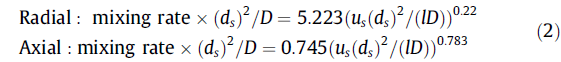
假设上述相关性关系适用于本系统,则可以研究段塞内的混合速率。考虑壁膜厚度,假设微批料的直径等于毛细管直径(= 0.5mm)。发现微批长度为0.7 mm,而混合物的扩散系数假定为10-8 m2/s,得到Pe* = 4244(速度= 0.12 m/s)。对于径向布置,发现混合率为1.31 s-1,而对于轴向布置,混合率为20.53 s-1。观察到的速率常数在0.038 ~6 s-1的范围内,这表明一些数据点具有混合影响,其对应于从(6)到(9)的变换(参见图5),即速率常数ki2,ob。使用更高的流速可以解决这个问题,然而,在高速下无法形成明显的团状流。因此,现已经发现动力学控制的五个步骤中的四个。特别是对于(1)-(3)的转化而言的低活化能是由于需要低能量的质子化。
将来,将建立速率常数kc2,ki2和ki3与研究反应动力学所需的平衡常数Kc和Ki之间的关系。
4.结论
在目前的工作中,微毛细管反应器中的团状流(微批)可用于研究快速且高放热反应的反应机理。该方法应用于代表环化反应类别的PI环化的模型反应。非常小的反应体积和非常短的反应时间允许抑制温度升高和捕获中间物质。在等温条件下研究了反应动力学,并提出了可靠的反应网络,以纠正现有的机理。开发的动力学模型与实验数据一致。最后,研究微批系统中的混合并鉴定动力学控制步骤。
所开发的方法具有通用性质,并且可以通过选择与参与反应的物质不混溶的合适的惰性载体流体而应用于各种快速放热的均相反应。
致谢
作者承认第7届欧洲框架计划PILLS项目的财政支持(拨款协议编号:CP-FP 214599)。
附录A.
考虑到图5所示方案的所有步骤为一阶反应,以及Kc和Ki表示的瞬时平衡,开发了质量平衡方程:
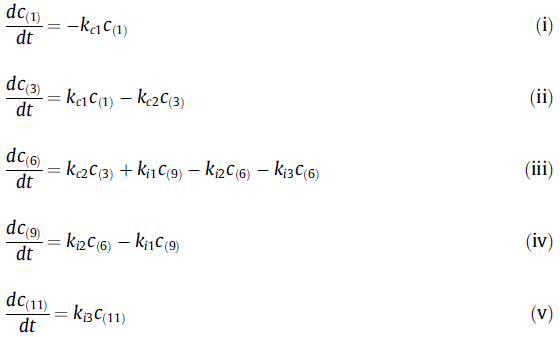
为了研究来自可测量化合物的反应动力学,需要建立“真实”和“观察”速率常数之间的关系。假设(3)和(4)以及(6)和(7)之间的瞬时平衡,这意味着测得的10-羟基衍生物和γ-紫罗兰酮分别为(3 + 4)和(6 + 7):

或者就(3)和(6)的浓度而言:

上述等式可以用平衡常数Kc= C(3)/C(4)和Ki = C(6)/ C(7)来表示,如下所示:

因此,由等式i,ii,iii,iv,v和等式(viii),我们得到:
其中,
使用商业软件Berkeley Madonna TM将上述方程组拟合到MSR实验数据,以确定五个速率常数。
参考文献
[1] R.L. Berger, Some problems concerning mixers and detectors forstopped flow kinetic studies, Biophys. J. 24 (1) (1978) 2–20.
[2] J.-I. Yoshida, Flash chemistry using electrochemical method andmicrosystems, Chem. Commun. 36 (2005) 4509–4516.
[3] P.B. Palde, T.F. Jamison, Safe and efficient tetrazolesynthesis in a continuousflow microreactor, Angew. Chem.-Int. Ed. 50 (15)(2011) 3525–3528.
[4] G. Dummann, U. Quittmann, L. Groschel, D.W. Agar, O. Worz, K. Morgenschweis,The capillary-microreactor: a new reactor concept for the intensification ofheat and mass transfer in liquid–liquid reactions, Catal. Today 79 (1–4) (2003)433–439.
[5] R. Abdallah, B. Fumey, V. Meille, C. de Bellefon, Micro-structuredreactors as a tool for chiral modifier screening in gas–liquid–solid asymmetrichydrogenations, Catal. Today 125 (1,2) (2007) 34–39.
[6] R. Abdallah, V. Meille, J. Shaw, D. Wenn, C. De Bellefon,Gas–liquid and gas– liquid–solid catalysis in a mesh microreactor, Chem.Commun. 10 (4) (2004) 372–373.
[7] T. Iwasaki, N. Kawano, J. Yoshida, Radical polymerization usingmicroflow system: numbering-up of microreactors and continuous operation, Org. Process.Res. Dev. 10 (6) (2006) 1126–1131.
[8] T. Iwasaki, J. Yoshida, Free radical polymerization inmicroreactors. Significant improvement in molecular weight distributioncontrol, Macromolecules 38 (4) (2005) 1159–1163.
[9] M.T. Janicke, H. Kestenbaum, U. Hagendorf, F. Schuth, M.Fichtner, K. Schubert, The controlled oxidation of hydrogen from an explosivemixture of gases using a microstructured reactor/heat exchanger and Pt/Al2O3catalyst, J. Catal. 191 (2) (2000) 282–293.
[10] K. Jähnisch, V. Hessel, H. Löwe, M. Baerns, Chemistry inmicrostructured reactors, Angew. Chem.-Int. Ed. 43 (4) (2004) 406–446.
[11] M. Ueno, H. Hisamoto, T. Kitamori, S. Kobayashi,Phase-transfer alkylation reactions using microreactors, Chem. Commun. 8 (2003)936–937.
[12] R.A. Maurya, C.P. Park, J.H. Lee, D.P. Kim, Continuous in situgeneration, separation, and reaction of diazomethane in a dual-channelmicroreactor, Angew. Chem.-Int. Ed. 50 (26) (2011) 5952–5955.
[13] T. Fukuyama, M.T. Rahman, M. Sato, I. Ryu, Adventures in innerspace. Microflow systems for practical organic synthesis, Synlett 2 (2008)151–163.
[14] K. Geyer, T. Gustafsson, P.H. Seeberger, Developingcontinuous-flow microreactors as tools for synthetic chemists, Synlett 15(2009) 1593–1612.
[15] V. Hessel, J.C. Schouten, A. Renken, J. Yoshida (Eds.), Handbookof Micro Reactors, Wiley-VCH, Weinheim, 2009.
[16] S.V. Luis, E. Garcia-Verdugo (Eds.), Chemical Reactions andProcesses Under Flow Conditions, Royal Society of Chemistry, 2009.
[17] C. Wiles, P. Watts, Continuous flow reactors, a tool for themodern synthetic chemist, Eur. J. Org. Chem. 2008 (10) (2008) 1655–1671.
[18] T. Wirth (Ed.), Microreactors in Organic Synthesis andCatalysis, Wiley-VCH, Weinheim, 2008.
[19] J.-i. Yoshida, A. Nagaki, T. Yamada, Flash chemistry: fastchemical synthesis by using microreactors, Chem. – A Eur. J. 14 (25) (2008)7450–7459.
[20] M. Kashid, A. Renken, L. Kiwi-Minsker, Mixing efficiency andenergy consumption for five generic microchannel designs, Chem. Eng. J. 167(2,3) (2011) 436–443.
[21] H. Song, R.F. Ismagilov, Millisecond kinetics on amicrofluidic chip using nanoliters of reagents, J. Am. Chem. Soc. 125 (47)(2003) 14613–14619.
[22] Z.T. Cygan, J.T. Cabral, K.L. Beers, E.J. Amis, Microfluidicplatform for the generation of organic-phase microreactors, Langmuir 21 (8)(2005) 3629–3634.
[23] B. Ahmed, D. Barrow, T. Wirth, Enhancement of reaction ratesby segmented fluid flow in capillary scale reactors, Adv. Synth. Catal. 348 (9)(2006) 1043–1048.
[24] C. de Bellefon, R. Abdallah, T. Lamouille, N. Pestre, S. Caravieilhes,P. Grenouillet, High-throughput screening of molecular catalysts using automatedliquid handling, injection, and microdevices, Chimia 56 (11) (2002) 621–626.
[25] M.N. Kashid, I. Yuranov, P. Raspail, P. Prechtl, J. Membrez,A. Renken, L. KiwiMinsker, Cyclisation of pseudoionone to b-Ionone: reactionmechanism and kinetics, Ind. Eng. Chem.Res. 50 (13) (2011) 7920–7926.
[26] J.R. Burns, C. Ramshaw, The intensification of rapid reactionsin multiphase systems using slug flow in capillaries, Lab on a Chip 1 (1)(2001) 10–15.
[27] J.D. Tice, H. Song, A.D. Lyon, R.F. Ismagilov, Formation ofdroplets and mixing in multiphase microfluidics at low values of the reynoldsand the capillary numbers, Langmuir 19 (22) (2003) 9127–9133.
[28] A.V. Semenovskii, V.A. Smith, V.F. Kucherov, The mechanism ofpseudoionone cyclisation, Dokl. Akad. Nauk SSSR+ 132 (5) (1960) 1107–1110.
[29] A.V. Semenovskii, V.A. Smith, V.F. Kucherov, The mechanism ofpseudoionone cyclisation, Dokl. Akad. Nauk SSSR+ 132 (5) (1960) 1107–1110.
[30] W. Tanthapanichakoon, K. Matsuyama, N. Aoki, K. Mae, Design ofmicrofluidic slug mixing based on the correlation between a dimensionlessmixing rate and a modified Peclet number, Chem. Eng. Sci. 61 (22) (2006)7386–7392.
[31] K. Matsuyama, W. Tanthapanichakoon, N. Aoki, K. Mae, Operationof microfluidic liquid slug formation and slug design for kinetics measurement,Chem. Eng. Sci. 62 (18–20) (2007) 5133–5136.
原文标题:《Micro-batch reactorfor catching intermediates and monitoring kinetics of rapid and exothermichomogeneous reactions》
原文出处:ChemicalEngineering Journal 214 (2013) 149–156
涉及β-紫罗兰酮的合成


目前评论: