- A+

吡啶是药物分子中普遍存在的结构单元。对临床候选药物中的吡啶单元进行化学修饰可以获得大量衍生物,这对新药研发过程十分重要。因此,开发取代吡啶的合成方法受到了研究人员的广泛关注。尽管Minisci反应是吡啶直接自由基烷基化的有力工具,但这类反应通常需要苛刻的条件(Trends Heterocycl. Chem. 2008, 13, 1–68)。近期,吡啶的光催化自由基烷基化取得了巨大的进展,反应在非常温和的条件下即可完成,但此类反应也面临着区域化学选择性难以控制的问题。
(Angew. Chem. Int. Ed. 2014, 53, 4802–4806; Nature 2015, 525, 87–90; J. Am. Chem. Soc. 2013, 135, 12122−12134)
吡啶N-氧化物是一类稳定的有机化合物,可作为起始原料合成吡啶衍生物。以储量丰富、容易获得的烯烃为烷基化试剂,实现吡啶N-氧化物到邻位烷基取代吡啶的转化,这种思路十分有吸引力。然而,这类报道仅有少数几例,而且只适用于缺电子烯烃。例如,全氟丙烯与吡啶N-氧化物通过1,3-偶极环加成和碳酰氟消除反应得到四氟乙基取代的吡啶(图1a,J. Org. Chem. 1968, 33, 3343–3344;Mendeleev Commun. 2006,
16, 161–163)。尽管吡啶N-氧化物中含有极性N–O键,但对于环加成反应来说,它们的1,3-偶极子活性较低,其原因可能是反应中两个底物的芳香性同时被破坏。解决这个问题的办法之一是以光催化简单烯烃生成的自由基物种作为吡啶N-氧化物的烷基化组分。因为这类阳离子自由基具有足够的亲电性,可与吡啶N-氧化物偶联生成自由基中间体,再经过分子内邻位加成和羰基化合物消除之后,便可得到邻位取代的吡啶产物。
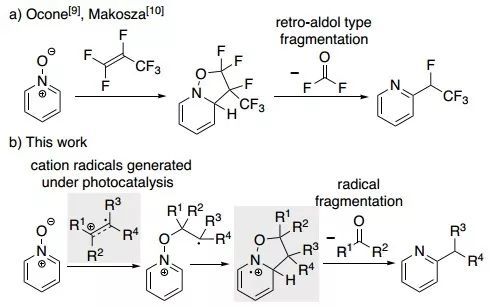
图1. 吡啶N-氧化物转化为邻位烷基取代吡啶
(来源:Angew. Chem. Int. Ed.)
近日,来自京都大学的Masahiro Murakami课题组报道了通过光催化烯烃裂解实现吡啶N-氧化物到邻位烷基取代吡啶的一步转化(图1b)。相关论文发表在Angew. Chem. Int. Ed.上(DOI: 10.1002/anie.201801305),通讯作者为村上正浩(Masahiro Murakami)教授和三浦智也(Tomoya Miura)副教授,论文一作为博士研究员周旺(Wang Zhou)。
研究初期,作者以丙酸肉桂酯(1a,0.50 mmol)和吡啶N-氧化物(2a,1.0 mmol)为反应底物,加入光催化剂Mes-Acr+ BF4–(5.0 mol %)和HBF4(42 %水溶液,85 mol %),在蓝色LED(446 nm,18.4 W)光照下反应6天,所得产物2-苄基吡啶(3aa)的分离产率为69%(图2,entry 1)。进一步实验表明,缩短反应时间以及把HBF4替换成其他酸,产率均下降;不添加HBF4时几乎没有目标产物生成。对照实验表明光催化剂和光照是反应进行的必要因素。此外,加入自由基捕获剂TEMPO反应不能发生,表明该反应是通过自由基机理进行的。
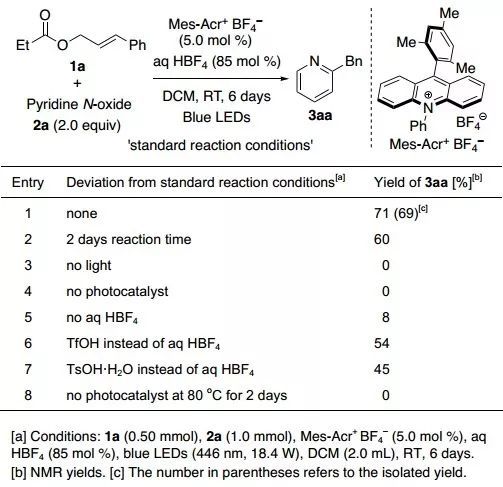
图2. 光催化丙酸肉桂酯吡啶N-氧化物的反应
(来源:Angew. Chem. Int. Ed.)
以上述反应为例,作者提出了这类反应的机理(图3)。首先,烯烃1a与激发态光催化剂PC*发生单电子转移,生成阳离子自由基I。然后,吡啶N-氧化物2a与I加成形成苄基自由基II,环化后转化为铵阳离子自由基III;或者2a与I直接环化生成III。接着,III经过去质子化和分子内1,2-单电子迁移转化成α-氨基自由基IV,并在芳构化驱使下经历β-N–O键断裂和质子化过程生成烷氧基自由基V。随后通过β-C–C断裂脱去醛部分构建苄基自由基VI,其可以接受一个电子实现光催化剂的翻转。最后,VI经过互变异构化释放出目标产物3aa。在反应过程中,亚化学计量的HBF4可以提供足够的酸性条件来保证质子化中间体V的生成。
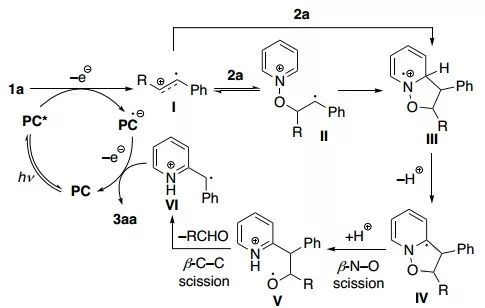
图3. 提出的机理
(来源:Angew. Chem. Int. Ed.)
最后,作者对反应底物吡啶N-氧化物和烯烃进行了拓展。一系列邻、间、对位被烷基或苯基取代的吡啶N-氧化物,以及喹啉、异喹啉、菲啶、苯并喹啉等氮杂稠环N-氧化物,都适用于这类反应条件(图4)。烯烃底物的适用范围同样很广泛,多种肉桂醇酯、链状烯烃、三取代烯烃等均能顺利反应。
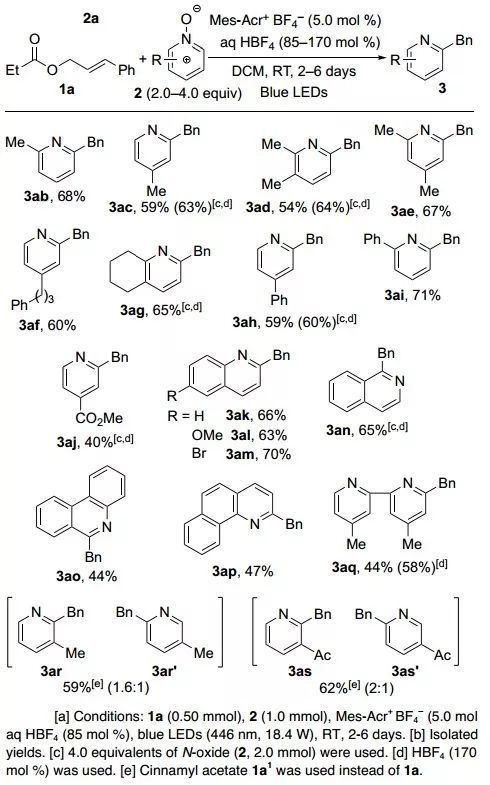
图4. 吡啶N-氧化物底物拓展
(来源:Angew. Chem. Int. Ed.)
总结:
Murakami课题组开发了以烯烃为烷基化试剂的光诱导吡啶N-氧化物的邻位烷基化反应。课题组将该策略成功应用到一系列吡啶N-氧化物和烯烃底物上,得到了邻位含有苄基和仲烷基的吡啶产物。


目前评论: