- A+
Carreira合成吲哚霉素B.
吲哚霉素家族的一些成员显示出有效的抗肿瘤活性。在到Indoxamycin B的路由密钥环戊烯形成工序(3)设计(Angew化学国际版。 2012,51,3474. DOI:10.1002 / ange.201109175)由苏黎世的埃里克M. Carreira的是钯- 催化环化1至2。
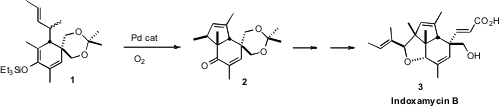
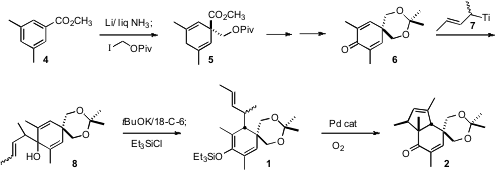
制备1的原料是对称的苯甲酸甲酯4。溶解金属还原,然后烷基化所得的酯烯醇化物,得到二烯5。还原和保护,然后将烯丙基氧化转化为5至6,其作为几何异构体的混合物进行至8。8的解离的醇钾经历平滑的氧 - Cope重排,得到被作为甲硅烷基醚1捕获的烯醇化物。Pd催化的氧化环化然后完成2的合成。
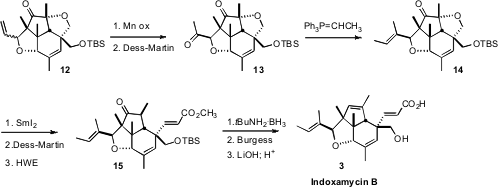
在手中存在酮2的情况下,仍存在两个挑战:区分两个羟甲基,并使烯丙基次甲基官能化以构建第三个季铵中心。这两个问题都通过V介导的对应于2的二醇的环氧化来解决。在原位,抗羟基开环氧化物,得到环醚。金 - 介导的O-炔丙基化烯醇醚10的重排递送了丙二烯,其被选择性地还原为醇11。第二次金介导的环化然后完成了12的合成。

已将吲哚霉素B指定为具有丁烯基侧链作为更稳定的内型非对映异构体,因此合成最初以这种方式完成。当该产品被证明与天然材料不一致时,似乎丁烯基侧链实际上是exo。12的选择性水合产生 所有四种醇非对映异构体的混合物。将两种外型非对映异构体分离并氧化成酮13。Wittig烯化产生一对几何异构体,将其分离。将E 异构体14的环醚还原,得到酮醇,将其氧化成酮醛。Horner-Wadsworth-Emmons链扩展得到15,这是继续使用Indoxamycin B(3)。
可以想象通过分子内卡宾插入2的烯丙基次甲基的3的替代构造。由北达科他州大学的克塞V. Novokov(开发的协议。四面体通讯2009年,50,6963, :DOI 10.1016 / j.tetlet.2009.09.147杂环; 2009,78,2531, DOI:10.3987 / COM-09例如,-11788)可以与对应于2的仲醇一起使用。


目前评论: