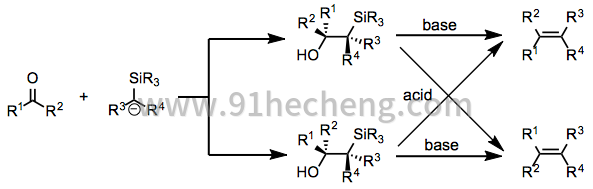
- A+
所述的McMurry coupling是二羰基官能团的反应以建立羰基的碳原子之间的新双键。该反应由低价钛试剂介导,其可以通过氯化钛与任何许多还原剂的组合产生。McMurry偶联可用于构建空间位阻烯烃,但由于缺乏立体化学控制和混合偶联反应中产物的统计混合物,因此范围有限。
介绍
1970年首次报道了芳香族羰基化合物与铝汞合金的频哪醇偶联中烯烃作为次要产物的形成。[2]从那时起,羰基化合物与烯烃的还原偶联已经发展成为一种有用的合成方法,最值得注意的是麦克默里和他的同事们。现代McMurry联轴器采用由钛源和还原剂(方程式1)产生的低价钛,并且反应的范围得益于几种钛还原剂组合的开发。醛和酮可以以分子内或分子间方式偶联,以提供可能难以使用其他方法获得的烯烃。在某些情况下,羧酸衍生物如酯,酰胺和硫酯易于偶联。
(1)

通常,产物的(E) - 异构体优于(Z) - 异构体,尽管当羰基的取代基的大小相似时可能产生混合物。已经假设单电子和双电子转移机制用于McMurry耦合,并且其机制的细节仍然是未知的。当所涉及的羰基的空间环境和还原电位相似时,实现选择性混合偶联(而不是同偶联和混合偶联产物的统计混合物)通常是困难的。避免这个问题的方法依赖于使用羰基等价物,例如硫代缩醛和偕二卤化物。
机制与立体化学
流行机制
McMurry耦合的机制目前尚不清楚,孤立的观察指出了单电子和双电子机制。在任何一种情况下,还原产生有机金属中间体的羰基是该方法的关键的第一步,并且可能随后进行脱氧。单电子还原可以提供两个钛羰基1,其随后可以彼此偶联以产生夹竹桃酸酯2。然后消除两个钛氧代分子,得到产物烯烃。这种机制很可能是脂肪族羰基化合物,并且已经被电子顺磁共振光谱学支持。[3]更容易还原的芳族羰基化合物经历双电子还原以产生钛羰基阴离子3,其在加入第二分子的羰基化合物(方程式2)后产生相同的pinacolate 2。[4]
(2)
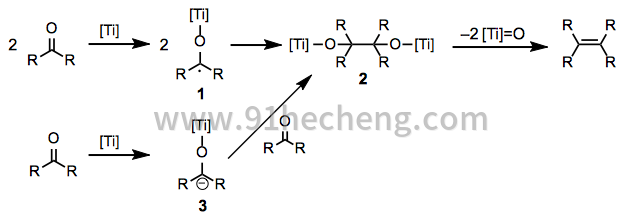
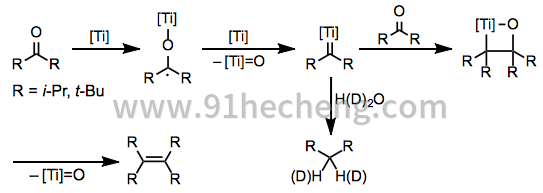
对于受阻酮,还发现了一种令人联想到涉及钛卡宾配合物的烯烃复分解的机理(方程式3)。[5] Pinacolate 2不是这些反应中的中间体 - 通过其他方式产生的相应钛的pinacolates分解为起始酮而不是烯烃。用水淬灭卡宾中间体得到相应的烷烃。
(3)
立体化学
的McMurry偶联通常产生的(混合物ë) - (和Ž) -异构体,与(ë) -异构体占统治地位。增加取代基之间的尺寸差异增加了对空间位阻较小的(E) - 异构体的选择性。[6]
(4)
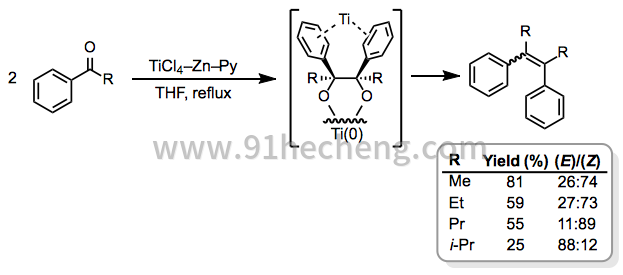
单芳基酮之间的偶联反应是该规则的有趣且重要的例外。当R较小时,这些反应产生(Z) - 异构体作为主要产物的趋势归因于芳基与钛中心的配位(方程式5)。[7]
(5)
范围和限制
采用钛的McMurry偶联需要低价物质,其通常通过用还原剂处理卤化钛而“原位”生成。已经使用各种钛还原剂组合进行反应,并且每种组合物具有独特的范围。还原剂包括锌金属,Zn / Cu,LiAlH 4,碱金属和碱土金属,锂芳烃和丁基锂。TiCl 4和TiCl 3是最常用的钛源。通常,脂族酮比芳族酮更难结合。例如,脂族酮在由TiCl 4和Zn 产生的低价钛试剂存在下专门形成频哪醇,并且是钛粉末的不良底物。
McMurry联轴器中使用的其他金属包括锆,钒,[8]铌,[9]钼,[10]钨,[11]铝,[12]和锌。[13]一些这些金属的实际效用由它们的成本和可用性的限制,但反应的范围从大量可使用的金属试剂的一定的好处。
在一些情况下,添加剂可以对由于频哪醇形成和重排导致产率降低的McMurry偶联物产生有益效果。例如,胺类抑制了频哪醇的形成和β-紫罗兰酮同源偶联中的重排(方程式6)。[14]亚化学计量的碘促进脂族羰基化合物在低温和短反应时间下通过TiCl 3 -Li 的偶联。[15]
(6)

羰基底物的范围受到低价钛试剂降低大量有机官能团的倾向的限制。例如,TiCl 3 -LiAlH 4体系将环氧化物和溴醇转化为烯烃并介导烯丙基和苄醇的脱氧。[16]羧酸底物通常在McMurry偶联中无效,但酯和酰胺的分子内混合偶联可用于制备杂环化合物(见下文)。
不饱和羰基化合物在McMurry偶联中平稳反应,在碳 - 碳双键处保留构型。[17]尽管两种酯之间的分子间耦合尚未报道,与其它羰基官能酯的混合耦合是已知的。在酮与酯的反应中,酮的均偶联可能是一个重要问题(方程式7)。[18]
(7)
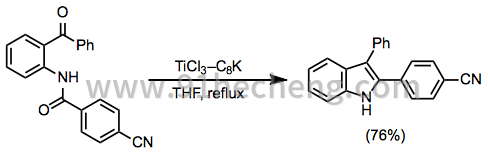
酰胺是相对通用的底物,并且已经报道了酰胺的分子间和分子内同型偶联反应。[19]酰胺与酮的分子内偶联已用于合成吲哚衍生物(方程式8)。[20]
(8)
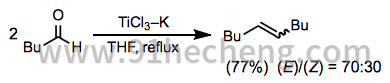
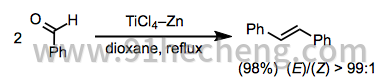
同源偶联反应是可以在McMurry偶联条件下完成的最直接的转换。脂肪族和芳香族酮可以高收率和立体选择性转化为相应的对称烯烃。在可能形成非对映异构体的反应中,(E) - 异构体的选择性是典型的(方程式9)。[3]
(9)
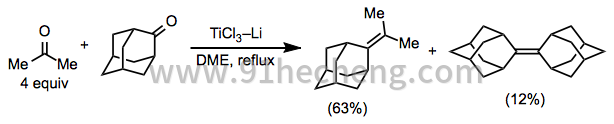
具有不同取代模式的羰基底物之间的混合偶联反应通常提供产物的统计混合物,除非使用过量的一个偶联配偶体(方程10)。[6]混合耦合的成功还取决于基板的结构; 在某些情况下,过量的偶联配偶之一不会使同源偶联最小化。
(10)
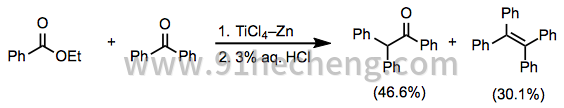
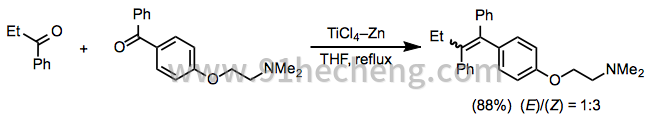
当所用两种基质的还原电位充分不同时,可以使用等摩尔量的两种偶联配偶实现选择性混合偶联。例如,在TiCl 3 -Zn 存在下,单芳基和二芳基酮易于以高收率彼此偶合(方程11)。[21]
(11)
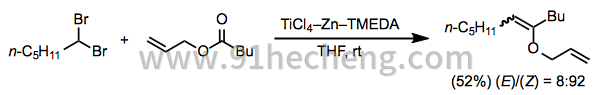
McMurry偶联受到以下缺点的严重限制:酮和醛之间的混合偶联难以实现。采用羰基等价物反应,如宝石 -dihalides和硫缩醛是适合于混合耦合,并很好地补充传统的McMurry偶联。基本上,这些反应是使用具有非常不同的还原电位的偶联伴侣的策略的延伸。酯,酰胺和硫酯是有用的底物并提供富含电子的烯烃(方程12)。[22]
(12)
合成应用
通常,McMurry偶联比合成的烯化方法更少用于合成 - 后期合成中间体中羰基的普遍存在和反应的还原条件限制了其对合成的适用性。它对于合成相当奇特的化合物非常有用,其中烯烃是关键断开的。例如,该反应可用于含有小的应变环的酮的烯化(方程13)。[12] [23]
(13)
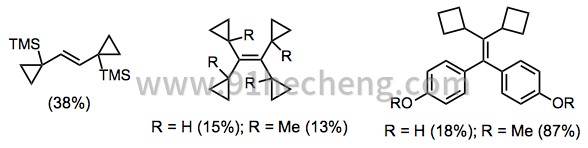
与需要形成闭合的四元中间体(例如Wittig和Horner-Wadsworth-Emmons反应)的烯化方法不同,McMurry偶联通过开放过渡态和中间体进行。因此,该反应可用于建立严重受阻的烯烃,其通过其他方式难以接近。[24]高度紧张的烯烃在学术意义上和高能量燃料中都很有趣。
与其他方法的比较
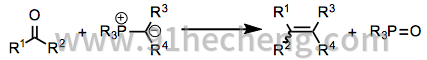
McMurry偶联受限于E / Z混合物的形成和在许多混合偶联反应中形成产物的统计混合物。存在许多替代的烯化方法,并且这些方法倾向于在有机合成中主导McMurry偶联。Wittig反应(方程14)使用羰基化合物与鏻叶立德(后者,在某种意义上,作为羰基当量)的组合。[25]
(14)
Peterson烯化反应依赖于α-甲硅烷基阴离子中间体,并且具有通过改变所用后处理可以获得双键异构体的优点。碱性后处理导致羟基和甲硅烷基的同步消除,而酸性后处理导致抗消除。[26]
(15)
涉及预形成或中间金属卡宾的羰基烯烃化试剂为McMurry偶联提供了另一种替代方案(方程16)。与高价金属如铌和钽相关的Schrock卡宾与羰基化合物反应得到相应的烯烃。[27]Tebbe试剂,一种钛亚甲基配合物,用于由醛和酮形成末端烯烃。[28]
(16)

实验条件和程序
典型条件
可以通过两种方式生成McMurry耦合所必需的低价钛物质。当使用的还原剂具有高反应性时,必须在加入羰基化合物之前产生试剂。然而,当基材对还原剂不敏感时,可以立即加入所有三种组分(钛源,还原剂和基质)。分子内偶联通常要求底物以非常低的浓度存在以避免寡聚化。钛源和还原剂的选择是关键的,因为不同的低价钛化合物是由不同的试剂组合产生的,并且这些化合物可能在特定的基质上起不同的作用。
在McMurry偶联剂中观察到有趣的溶剂对产率和立体选择性的影响,[29]但反应通常在四氢呋喃,二恶烷或其他醚溶剂中进行。通常需要升高的温度以避免分离频哪醇,因为反应的主要产物 - 热促进了山椒酸钛中间体的脱氧。
示例程序[30]
(17)
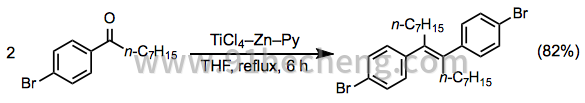
在-5至5℃,N 2下,将TiCl 4(19.0g,0.1mol)缓慢加入无水THF(400mL)中。向得到的黄色溶液中加入锌(18.0g,0.27mol)。然后通过注射器注射吡啶(3mL)。将反应混合物加热至回流,然后在1小时内加入酮(37.0g,0.13mol)的THF(100mL)溶液,并将混合物再回流5小时。冷却后,减压除去溶剂。剩余的深色固体用饱和K 2 CO 3水溶液处理溶液并搅拌过夜。过滤除去固体,在70℃下减压干燥,用沸石轻石油醚进行索氏提取3天。浓缩萃取物,并通过硅胶色谱(CH 2 Cl 2)纯化残余物。静置时结晶的纯产物(28.4g,82%):无色晶体,熔点103℃; 1 H NMR(200MHz,CDCl 3)δ0.80(t,6H,CH),1.10-1.38(m,20H,CH),2.45(m,4H,α-CH 2),6.77,7.17(AA'BB) ',4H,ArH); 13 C NMR(50MHz,CDCl 3)δ14.1,22.6,23.6,28.3,29.1,31.8,34.2,119.5,137.9,130.7,131.3,142.0。肛门。C 28 H 计算值38 Br 2:C,62.93; H,7.17; Br,29.90。实测值:C,63.23; H,7.38; Br 29.25。


目前评论: