- A+
亲核环氧化是通过亲核氧化剂的作用从缺电子双键形成环氧化物。亲核环氧化方法代表了亲电子方法的可行替代方法,其中许多方法不能有效地使贫电子双键环氧化。
介绍
尽管最常用的不对称环氧化方法(Sharpless-Katsuki,[2]和Jacobsen [3]环氧化物)依赖于亲电子氧化剂的催化反应性,但用合适的离去基团取代的亲核氧源也可用作环氧化试剂。典型的例子,Weitz-Scheffer反应[4]在碱性条件下使用过氧化氢(Z = OH以下)。其他值得注意的例子使用了次氯酸盐(Z = Cl)和手性过氧化物(Z = OR *)。
(1)
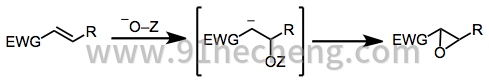
上述反应的不对称形式利用了许多实现不对称诱导的策略。最高产和最具对映选择性的方法包括:
使用化学计量手性氧化剂[5]
使用被手性配体取代的化学计量金属过氧化物[6]
使用化学计量手性碱[7]
多肽的使用[8]
尽管这些反应中的每一种的机制都有所不同,但在每种情况下,手性催化剂或试剂必须参与对映异构化的缀合物添加步骤。应当指出的是,顺式 -环氧化物很难用亲核环氧化的方法来访问,几乎所有的亲核环氧化顺烯烃不起反式环氧化物。
机制与立体化学
流行机制
亲核环氧化的机理始于将过氧化物(或其他O-亲核物质)共轭加成到烯酮上。溶液中存在的金属离子或共轭酸协调过氧化物氧和烯醇化氧。烯醇化物对过氧化物氧的攻击产生环氧化物产物并释放离去基团。
(2)
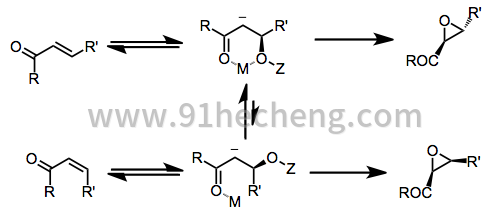
因为该过程是逐步的,所以不一定保留碳 - 碳双键的构型。无论顺和反式烯酮形成反式环氧化物几乎所有的亲核环氧化条件下(方法采用镧系元素的BINOL系统除外)。
立体选择性变体
在不对称亲核环氧化中如何实现立体选择性取决于所采用的方法。这里涵盖了各种用于贫电子烯烃的不对称亲核环氧化的方法。请参阅下文,了解反应的底物范围。
当使用手性非外消旋过氧化物时,导致对映体产物的环氧化的两种过渡态是非对映异构体。过氧化物,烯酮和模板阳离子M +之间的空间相互作用影响观察到的选择性。[9]
(3)
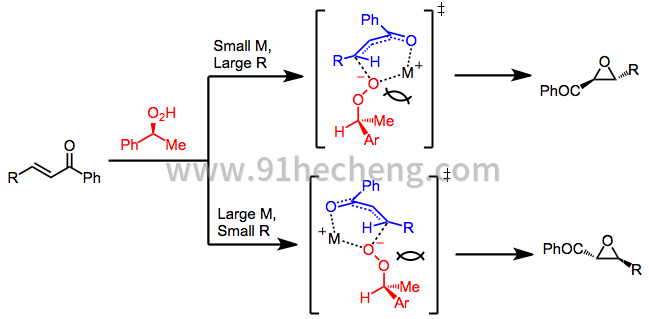
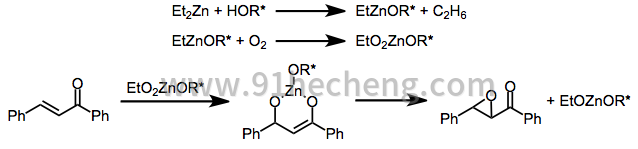
采用由手性非外消旋配体改性的金属过氧化物的方法通过类似的机理起作用,其中金属阳离子起模板作用。在氧气氛下的手性锌醇盐已被用于环氧化某些类别的烯酮(参见下面的等式(8))。乙烷气体的释放和氧气的吸收是配体交换的证据,随后是中间体锌醇盐物质的氧化。[10]
(4)
锂,镁和钙[11]烷基过氧化物也已用作不对称亲核环氧化试剂。简单的酒石酸盐和伪麻黄碱配体与这些金属结合使用是有效的; 然而,关于这些系统的精确机制的详细信息很少是已知的。
与BINOL配体和氢过氧化枯烯组合,镧系元素醇盐可用于使反式和顺式烯酮环氧化,具有高对映选择性。用这些催化剂体系研究非线性效应表明活性催化剂是低聚的。[12]
(5)
氨基酸(多肽)的均聚物也可用于在烯酮和过氧化物存在下进行对映选择性环氧化。结构 - 反应性关系尚未出现,但这些反应中的对映选择性通常很高,并且当其他方法失败时通常可以使用多肽。[13]
使用基于金鸡纳的生物碱催化剂也可以进行亲核环氧化的相转移催化。相转移方法允许所用氧化剂的一些变化:氢过氧化物,过氧化氢和次氯酸盐都已成功使用。[14]
范围和限制
对映选择性亲核环氧化的最佳条件取决于所用的底物。尽管可以使用亲核方法将各种基材环氧化,但是每种特定方法往往具有有限的基材范围。该部分描述了不对称亲核环氧化方法,根据不饱和底物的构成和构型组织它们。
烯酮
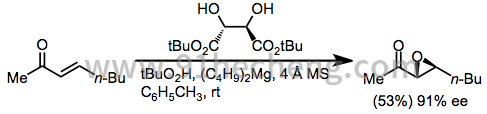
二烷基(E) - 烯酮最常使用镧系元素/ BINOL系统[15]或酒石酸镁催化剂[16]进行环氧化。
(6)
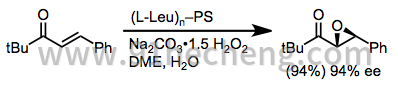
对于烷基芳基(E) - 烯酮,多肽[17]和镧系元素/ BINOL催化剂[18]都提供了良好的产率和对映选择性。最常用的多肽是聚-L-亮氨酸。
(7)
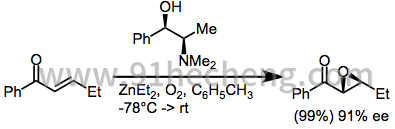
使用化学计量的过氧化锌体系,芳基烷基(E) - 烯酮已经以高对映选择性进行环氧化。[19]聚亮氨酸也可以与这些基质一起使用; [17]当底物中存在的立体中心偏向环氧化的选择性时,聚亮氨酸能够克服这种偏差。
(8)
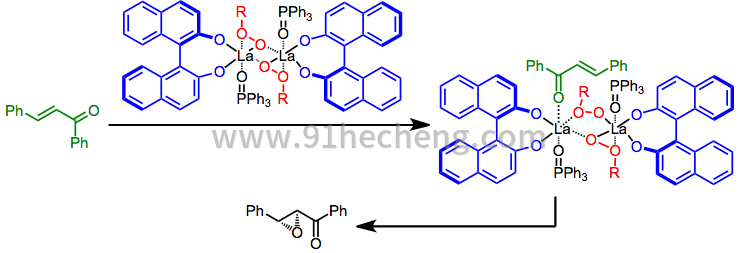
相转移催化已成功应用于二芳基(E) - 烯酮(查耳酮)的环氧化[14]。镧系元素/ BINOL对这类基质也是有效的。[20]
(9)
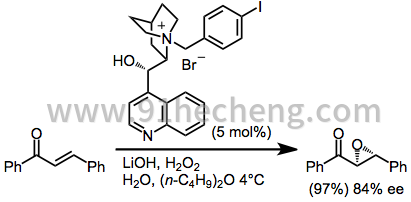
(Z) - 在没有中间键旋转的情况下难以环氧化以提供反式环氧化物。镧系元素催化剂确实有效地防止了键的旋转[18],并提供了顺式环氧化物产物。
(10)
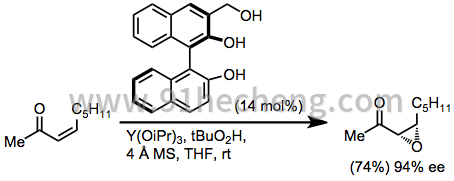
除了亚甲基四氢萘酮底物之外,[21]没有一般方法可用于三取代双键的不对称亲核环氧化。
其他电子贫乏的烯烃
可以使用亲电或亲核方法将不饱和酯环氧化。镧系元素介导的环氧化已成功应用于肉桂酸酯和β-杂芳基不饱和酯。[22]酰胺也在镧系元素介导的条件下被环氧化。[23]
(11)
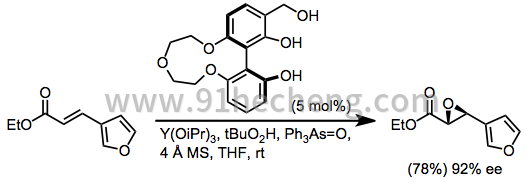
其他缺电子双键的环氧化(被除羰基以外的吸电子基团取代)的范围有限,尽管已经报道了一些例子。[24] [25]羰基配位路易斯酸性官能团的能力对于大多数现有方法至关重要。
与其他方法比较
醛和(α-) - 卤代酯之间的不对称Darzens反应是合成缩水甘油酯的有效方法。[26]手性助剂,[27]手性硼烯醇化物[28]和不对称相转移催化[29]已成功用于实现Darzens反应中的不对称诱导。
(12)
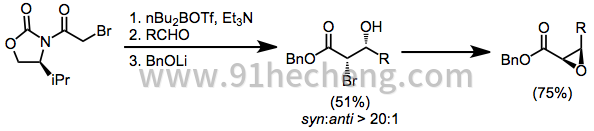
手性非外消旋烯烃的非对映选择性环氧化受到限制,即在不干扰环氧化物的情况下除去助剂通常是困难的。尽管如此,在某些情况下,非对映选择性很高。[30]
(13)
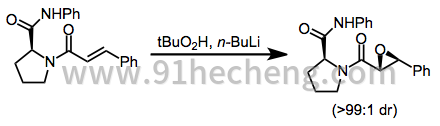
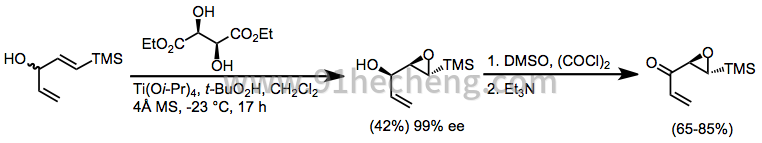
通过Sharpless环氧化产生的环氧醇的氧化是对映选择性合成手性α,β-环氧羰基化合物的第三种方法。[31] Swern和Parikh-Doering条件最常用于完成这些氧化。
(14)
实验条件和程序
典型条件
通常,亲核环氧化在惰性气氛下在无水条件下进行。对于锌介导的环氧化,首先将二乙基锌和配体混合并氧化,然后引入烯酮。镧系元素介导的环氧化通常需要添加剂来稳定催化剂; 这通常是三苯基氧化膦或三苯基胂氧化物。
相转移催化的环氧化可以使用三种可能的反应条件之一进行:(1)室温下的次氯酸钠,(2)新制备的8M次氯酸钾,或(3)水或非水中的三氯异氰尿酸条件。
在基于多肽的方法中,使用相转移催化剂和三相介质允许较低的催化剂负载量。也可以使用有机碱与脲/ H 2 O 2一起使用的双相条件。
示例程序[32]
(15)
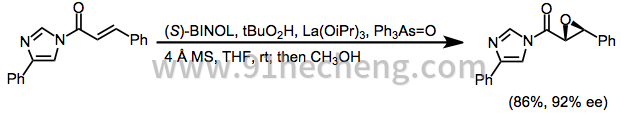
La(Oi-Pr)3的溶液(在THF中的0.2M,125μL,0.025mmol)加入到分子筛(250mg),(S)-BINOL(7.2mg,0.025mmol)和三苯基胂氧化物(8.1mg,0.025mmol)的悬浮液中。在室温下在THF(2.5mL)中。将溶液搅拌45分钟后,加入TBHP(5M癸烷,120μL,0.6mmol),将混合物在室温下再搅拌10分钟。向该溶液中加入(E)-3-苯基-1-(4-苯基咪唑-1-基) - 丙-2-烯-1-酮(68.6mg,0.25mmol),并将反应混合物在室温下搅拌温度1小时。加入甲醇(0.5mL),将混合物搅拌3小时,然后用0%的1%柠檬酸水溶液(2.5mL)处理。将混合物用EtOAc(2×10mL)萃取,并将合并的有机层用2%硫代硫酸钠水溶液(5mL)和盐水(5mL)洗涤,干燥(Na)。2 SO 4),减压除去溶剂。通过快速色谱(SiO 2; 50:1己烷/ EtOAc)纯化残余物,得到(2R,3S) - 2,3-环氧-3-苯基丙酸甲酯(38.5mg,86%),92%ee(通过HPLC测定; Daicel Chiralpak AD,98:2己烷/ i-PrOH),为无色油状物:[α] D(c 1.17,CHCl 3); 1 H NMR(400MHz,CDCl 3)δ3.52(d,J = 1.8Hz,1H),3.83(s,3H),4.10(d,J = 1.8Hz,1H),7.26-7.30(m,2H) ,7.35-7.39(m,3H)。


目前评论: